yupenghe / methylpy Goto Github PK
View Code? Open in Web Editor NEWWGBS/NOMe-seq Data Processing & Differential Methylation Analysis
License: Apache License 2.0
WGBS/NOMe-seq Data Processing & Differential Methylation Analysis
License: Apache License 2.0
I am trying to convert allc files to bigwig but I am getting some errors. Here is the command I ran:
$methylpy allc-to-bigwig --allc-file wtCol_R1_A_s.tsv --output wtCol_R1_A_s_CHH.bw --ref-fasta ../../../data/ref_annot/TAIR10.fasta --mc-type CHH --bin-size 100
The wigToBigWig script is installed in my PATH, I also ran methylpy by specifying the path, but I get the same error.
##########
Traceback (most recent call last):
File "/Users/xxxx/anaconda3/bin/methylpy", line 4, in <module>
__import__('pkg_resources').run_script('methylpy==1.3.1', 'methylpy')
File "/Users/xxxx/anaconda3/lib/python3.6/site-packages/pkg_resources/__init__.py", line 748, in run_script
self.require(requires)[0].run_script(script_name, ns)
File "/Users/xxxx/anaconda3/lib/python3.6/site-packages/pkg_resources/__init__.py", line 1517, in run_script
exec(code, namespace, namespace)
File "/Users/xxxx/anaconda3/lib/python3.6/site-packages/methylpy-1.3.1-py3.6.egg/EGG-INFO/scripts/methylpy", line 5, in <module>
parse_args()
File "/Users/xxxx/anaconda3/lib/python3.6/site-packages/methylpy-1.3.1-py3.6.egg/methylpy/parser.py", line 313, in parse_args
add_chr_prefix=args.add_chr_prefix)
File "/Users/xxxx/anaconda3/lib/python3.6/site-packages/methylpy-1.3.1-py3.6.egg/methylpy/utilities.py", line 252, in convert_allc_to_bigwig
+output_file),stderr=subprocess.PIPE)
File "/Users/xxxx/anaconda3/lib/python3.6/subprocess.py", line 291, in check_call
raise CalledProcessError(retcode, cmd)
subprocess.CalledProcessError: Command '['wigToBigWig', 'wtCol_R1_A_s_CHH.bw.wig', 'wtCol_R1_A_s_CHH.bw.chrom_size', 'wtCol_R1_A_s_CHH.bw']' returned non-zero exit status 255.
Hi Yupeng,
I was using add-methylation-level to summarize data to gene level. However, when I run the function, it seems something wrong with the output file.
The problem here is that the aggregated methylation data for each sample is not correctly aligned to match the genomic variables. In other words, I have 4 variables from genomic file, and 1 sample, so the output file is supposed to have 5 columns, but I only got 4 columns, and R can not read the sample column. It seems the methylation sample column was place to the next row instead of the same row. May I have your suggestion on dealing with it?

Thanks in advance and looking forward to your reply!
Hi Yupeng,
I am facing a problem with methylpy DMRfind. Everything looks good when I run the run_test.py :
Tue Nov 12 15:47:14 2019
Test importing methylpy module.
- importing call_mc_se: pass
- importing call_mc_pe: pass
- importing DMRfind: pass
- importing parser: pass
Check methylpy executable:pass
methylpy is successfully installed!
Check whether dependencies are available in PATH
- find bowtie: failed
- find bowtie2: found version 2.2.6
- find minimap2: found version 2.16-r922
- find samtools: found version 1.9
- find cutadapt: found version 2.6
- find java: found version 2019-03-19
Only bowtie2 is available. Skip tests for bowtie related code
Test DMRfind: pass
Test reidentify-DMR: pass
Test build-reference with bowtie2: pass
Test build-reference with minimap2: pass
Test single-end-pipeline with bowtie2: pass
Test single-end-pipeline with minimap2: pass
Test paired-end-pipeline with bowtie2: pass
Test paired-end-pipeline with minimap2: pass
Test quality filter for BAM file of single-end data: pass
Test call-methylation-state for single-end data: pass
Test call-methylation-state for paired-end data: pass
Test merge-allc: pass
Test filter-allc: pass
All tests are done!
However, when I try to use DMRfindon my data, I get the following:
/Library/Python/2.7/site-packages/methylpy/utilities.py:14: UserWarning: Module argparse was already imported from /System/Library/Frameworks/Python.framework/Versions/2.7/lib/python2.7/argparse.pyc, but /Library/Python/2.7/site-packages is being added to sys.path
from pkg_resources import parse_version
Filtering allc files using 2 node(s).
Tue Nov 12 15:44:40 2019
Splitting allc files for chromosome 1
Tue Nov 12 15:45:54 2019
(<type 'exceptions.KeyError'>, 184)
'1'
Running RMS tests failed.
May you please help me?
Hi Yupeng! How are you?! I'm excited to be using methylpy for a BS-seq analysis on Geoduck clams. I already had Bismark .bam files, and wanted to use methylpy to call DMRs. Because our samples have low coverage, I am wondering if there is an option to "destrand" like there is in Methylkit as part of the unite function (https://bioconductor.org/packages/release/bioc/vignettes/methylKit/inst/doc/methylKit.html). Hope all is going well in CA!
Hi,
There is a parameter for the adapter sequence as input, but unfortunately it is not working. If I use cutadapt to remove the adapters first and then input to methylpy, the mapping is much better. Any chance the adapter sequence is hardcoded into the program or am I running the program incorrectly. The following command did not successfully clip the adapters before mapping:
methylpy single-end-pipeline
--adapter-seq CTGTCTCTTATACACATCT
--read-files raw_reads/read.fastq.gz
--sample sampleName
--forward-ref index_methylpy_f
--reverse-ref index_methylpy_r
--ref-fasta template.fa
--num-procs 4
--unmethylated-control Control:
--binom-test TRUE
--min-cov 3
--remove-chr-prefix False
--pbat True
--compress-output FALSE
--path-to-output output/
Thanks for your help.
Can you provide sample data for the SNP calling function? I'd like to call all variants across all known SNPs and I'm not quite sure how. Since the bisulfite process actually seems to work by corrupting one of the strands, I'm nervous about using "normal" WGBS-unaware tooling.
Hi Yupeng,
I am using linux to run methylpy. I have problem with the first step building references. when I run the following script I get IOError: [Errno 71] Protocol error error:
methylpy build-reference
--input-files wtMouseTGFBI.fa
--output-prefix wtMouseTGFBI
--aligner minimap2
would you please let me know what the problem is? should I run this with python in linux? or just linux?
Thanks
Hi,
Merging of allc files doesnt sort the generated final allc file, will that be a problem during DMR calling? Also can you add an option to run binomial test for the generated file?
Rahul
Hi @yupenghe , happy Friday! I'm wondering if DMRfind calculates statistics to assess methylation differences between experimental groups ("sample-categories") or if it only calculates within-sample statistics. I'm wondering if I need to run an additional statistical test to identify group differences in % methylation from my rms_results_collapsed.tsv file.
Hi Yupeng! Is there rationale behind the default DMRfind --dmr_max_dist being 250bp?
The program automatically throws away multi-mapped reads and analyzes unique reads. Is there an easy way to allow it to process multi-mapped reads?
Hi yupenghe,
Can you look into the bam-quality-filter program? It does not function as its supposed to. I am trying to use it to filter reads with very high non-CG methylation >90%. The program is able to filter a subset of these reads successfully but not all of the highly methylated reads. In comparison, bismark's filte program is effectively able to remove all reads, but it needs initial mapping to be done in bismark and therefore I cannot use it for bam file generated through methylpy.
Thank you.
Hey Yupeng, I have run into some issues while running a full DMRfind pipeline. Could you suggest if the errors are due to the options I used in the following command?
$methylpy DMRfind --allc-files A.tsv .B.tsv --samples A B --chroms 1 2 3 4 5 --mc-type CHH --output-prefix AB --resid-cutoff 2 --min-num-dms 4
At the FDR correction stage, I get the following error messages:
Begin FDR Correction
Mon May 21 16:13:09 2018
m0 estimate for iteration 0: 3819203
Histogram FDR correction did not converge. Using the smallest p-value 0.0003333333333333333 as cutoff.
Calculating Residual Cutoff
Mon May 21 16:13:11 2018
Traceback (most recent call last):
File "/Users/x/anaconda3/bin/methylpy", line 4, in <module>
__import__('pkg_resources').run_script('methylpy==1.3.2', 'methylpy')
File "/Users/x/anaconda3/lib/python3.6/site-packages/pkg_resources/__init__.py", line 748, in run_script
self.require(requires)[0].run_script(script_name, ns)
File "/Users/x/anaconda3/lib/python3.6/site-packages/pkg_resources/__init__.py", line 1517, in run_script
exec(code, namespace, namespace)
File "/Users/x/anaconda3/lib/python3.6/site-packages/methylpy-1.3.2-py3.6.egg/EGG-INFO/scripts/methylpy", line 5, in <module>
parse_args()
File "/Users/x/anaconda3/lib/python3.6/site-packages/methylpy-1.3.2-py3.6.egg/methylpy/parser.py", line 71, in parse_args
seed=args.seed)
File "/Users/x/anaconda3/lib/python3.6/site-packages/methylpy-1.3.2-py3.6.egg/methylpy/DMRfind.py", line 274, in DMRfind
convergence_diff=convergence_diff)
File "/Users/x/anaconda3/lib/python3.6/site-packages/methylpy-1.3.2-py3.6.egg/methylpy/DMRfind.py", line 319, in merge_DMS_to_DMR
input_rms_file)
File "/Users/x/anaconda3/lib/python3.6/site-packages/methylpy-1.3.2-py3.6.egg/methylpy/DMRfind.py", line 487, in get_resid_cutoff
return scoreatpercentile(residuals, resid_cutoff)
File "/Users/x/anaconda3/lib/python3.6/site-packages/scipy/stats/stats.py", line 1590, in scoreatpercentile
return _compute_qth_percentile(sorted, per, interpolation_method, axis)
File "/Users/x/anaconda3/lib/python3.6/site-packages/scipy/stats/stats.py", line 1601, in _compute_qth_percentile
raise ValueError("percentile must be in the range [0, 100]")
ValueError: percentile must be in the range [0, 100]
Hello Yupeng,
Can you set TMP DIR option in remove_clonal_bam function? It is raising some writing errors.
something like this since TMPDIR can be set by user easily.
-Djava.io.tmpdir=$TMPDIR
Rahul
Is it just suit for WGBS of single cell ?
methylpy/methylpy/utilities.py
Line 116 in 5d43837
Hi Yupeng,
I am running methylpy on sample and the way methylation calling seems a bit random (in the table below check positions Chr1:102 and Chr1:107. Both are covered by only 1 read but still they are counted as methylated.)
Chr1 101 + CCT 0 1 0
Chr1 102 + CTA 1 1 1
Chr1 107 + CCC 1 1 0
Chr1 108 + CCG 1 1 1
Chr1 109 + CGA 1 2 1
Chr1 110 - CGG 5 5 1
Chr1 114 + CCG 2 2 1
Chr1 115 + CGG 2 2 1
Chr1 116 - CGG 6 7 1
Chr1 117 - CCG 1 7 0
The methylpy version I am using is 1.1.9, is it resolved in the newer versions?
Rahul
Is the index files are a set of files with the end of .bt2 ???
hello im having this issue.
[bam_sort_core] merging from 0 files and 8 in-memory blocks...
Error: Unable to access jarfile picard/picard.jar
Traceback (most recent call last):
File "/home/pocho/anaconda3/envs/BIT/bin/methylpy", line 5, in
parse_args()
File "/home/pocho/anaconda3/envs/BIT/lib/python3.7/site-packages/methylpy/parser.py", line 185, in parse_args
keep_temp_files=args.keep_temp_files)
File "/home/pocho/anaconda3/envs/BIT/lib/python3.7/site-packages/methylpy/call_mc_pe.py", line 276, in run_methylation_pipeline_pe
java_options=java_options)
File "/home/pocho/anaconda3/envs/BIT/lib/python3.7/site-packages/methylpy/call_mc_se.py", line 1342, in remove_clonal_bam
"QUIET=true"])
File "/home/pocho/anaconda3/envs/BIT/lib/python3.7/subprocess.py", line 363, in check_call
raise CalledProcessError(retcode, cmd)
subprocess.CalledProcessError: Command '['java', '-Xmx20g', '-jar', 'picard/picard.jar', 'MarkDuplicates', 'INPUT=result_pend/p_end_libA_processed_reads.bam', 'OUTPUT=result_pend/p_end_libA_processed_reads_no_clonal.bam', 'ASSUME_SORTED=true', 'REMOVE_DUPLICATES=true', 'METRICS_FILE=result_pend/p_end_libA.metric', 'VALIDATION_STRINGENCY=LENIENT', 'QUIET=true']' returned non-zero exit status 1.
i have already added java path to enviroment variable and have the path to picard.jar
any idea what could be going on ?
Hi! yupenghe,
I used methylpy which generated a bam file along with allc files. I need your suggestion regarding allc file contains all methylation context. I need to make separate files for CG, CHH, and CHG context and want to count (number as well in %) them individually.
Is there any way I can pull out all the context individually and count them as there is no separate file generated given details of methylation statics.
Thank you!
Best
CS791
Hi Yupeng,
When I ran the DMRfind, I got such error information.

The code I used is:
methylpy DMRfind
--allc-files ./Allc/ICE2-CR-1.tsv.gz ./Allc/ICE2-CR-3.tsv.gz ./Allc/ICE2-HR-3.tsv.gz
--samples CR-1 HR-3 CR-3
--mc-type "CGN"
--num-procs 8
--output-prefix ICE
And I found that gsl-2.5 didn't generate the file named libgsl.so.0. Only gsl-1.6 could generate libgsl.so.0. So could you adjust methlpy to fit both gsl 1.6 version and 2.5 verson?
Hey there,
while running the Methylpy tutorial I noticed that some of the commands need to replaced to accomodate changes introduced in the latest version.
Specifically, change methylpy build-reference options " --bowtie2 True" and "--parallel True"
Best
Christoph
Hi Yupeng,
Hope this finds you well!
I am now trying to use methylpy to summarize data into gene level by function "add-methylation-level". In other words, to generate a gene by cell matrix. However, I encountered a small problem, could you please be so kind as to do me a favor looking at it?
I inputted gene information to be the "input-tsv-file". For the input allc file, raw data is not available, so I am afraid single/paired-end-pipeline can not applied to generate allc file. What I could get is just 4 columns (i.e. chromosome, position, number of methylated reads and number of total reads). The remaining 3 columns (strand, sequence context, methylated) were missing in my case. Is it possible to run the "add-methylation-level" to get a gene by cell matrix?
Just for your information, I actually have used these 4 columns to run "add-methylation-level", but it turned out all the entries were "NA". I am quite confused about the result, and I guess it is because of the allc file.
Thanks in advance for your time! Looking forward to hear from you.
Hi Yupeng, I am now trying to compile the rms.cpp but it's not working in my system, could you help to figure out the problem? I installed gsl in /usr/local/, when I run the code provided in the tutorial, it could not locate gsl:
g++ -O3 -l gsl -l gslcblas -o run_rms_tests.out rms.cpp
rms.cpp:17:10: fatal error: 'gsl/gsl_rng.h' file not found
#include <gsl/gsl_rng.h>
^~~~~~~~~~~~~~~
1 error generated.
Then I tried specifying the path for gsl header files, but this produces another error:
$g++ -O3 -I /usr/local/include -L /usr/local/lib -l gsl -l gslcblas -o run_rms_tests.out rms.cpp
rms.cpp:88:1: warning: control reaches end of non-void function [-Wreturn-type]
}
^
1 warning generated.
Dear Yupeng,
I am having trouble with running DMRfind for some of my datasets that were downloaded from NCBI (Arabidopsis bisulfite seq GSE67216). I used methylpy to generate the allC files and then merge replicates for samples -all of which worked fine. When I run DMRfind, I get an error during residual cutoff stage (see below), and the final DMS/DMR files are not generated. Could you help me to figure out how to fix this?
Calculating Residual Cutoff
Tue Feb 18 11:16:32 2020
Traceback (most recent call last):
File "/home/s/miniconda3/bin/methylpy", line 5, in <module>
parse_args()
File "/home/s/miniconda3/lib/python3.7/site-packages/methylpy/parser.py", line 71, in parse_args
seed=args.seed)
File "/home/s/miniconda3/lib/python3.7/site-packages/methylpy/DMRfind.py", line 274, in DMRfind
convergence_diff=convergence_diff)
File "/home/s/miniconda3/lib/python3.7/site-packages/methylpy/DMRfind.py", line 319, in merge_DMS_to_DMR
input_rms_file)
File "/home/s/miniconda3/lib/python3.7/site-packages/methylpy/DMRfind.py", line 482, in get_resid_cutoff
residuals.append(float(resid))
ValueError: could not convert string to float: '-'
Here is the code I used for DMRfind
methylpy DMRfind --allc-files methylpy_results/merged_allc/allc_merged_A.tsv.gz methylpy_results/merged_allc/allc_merged_B.tsv.gz methylpy_results/merged_allc/allc_merged_C.tsv.gz --mc-type "CHH" --num-procs 8 --min-cov 3 --output-prefix repeat_dmrs/CHH_sig01_dms0_cov3
I have used the same code for other files and it works fine. The merged allC files don't seem to have any issue. Here are the first few lines from one of the allC files:
$gunzip -d -c allc_merged_A.tsv.gz |tail
3 23459756 + CAC 1 4 1
3 23459758 + CCT 1 4 1
3 23459759 + CTT 0 4 1
3 23459762 + CGA 0 3 1
3 23459763 - CGA 0 1 1
3 23459765 + CAC 0 3 1
3 23459767 + CCA 0 3 1
3 23459768 + CAG 0 2 1
3 23459770 - CTG 0 1 1
3 23459771 + CCT 0 1 1
$ gunzip -d -c allc_merged_A.tsv.gz |cut -f1|sort|uniq -c
10272463 1
6721681 2
8173516 3
6428501 4
9248527 5
42315 chloroplast
62756 mitochondria
Below are the codes to generate the allC files:
methylpy single-end-pipeline \
--read-files SRR1927188_GSM1642569.fastq.gz \
--sample rep1_SRR1927188 \
--trim-reads True \
--unmethylated-control chloroplast: \
--forward-ref TAIR10_chr_all.fas.shIDs_f \
--reverse-ref TAIR10_chr_all.fas.shIDs_r \
--ref-fasta TAIR10_chr_all.fas.shIDs.fa \
--num-procs 8 \
--compress-output False \
--path-to-output methylpy_results/
methylpy merge-allc \
--allc-files methylpy_results/allc_rep1_SRR1927188.tsv methylpy_results/allc_rep2_SRR1927189.tsv methylpy_results/allc_rep3_SRR1927190.tsv \
--output-file methylpy_results/merged_allc/allc_merged_C.tsv.gz \
--compress-output True
Hi @yupenghe ! In looking through DMRs and DMS in IGV, I'm finding some regions that look like they should be called DMRs, but there were not (see screenshot example). In the screenshot, the top 4 samples are one treatment group (orange), the next 4 are one treatment group (blue), and the last 4 are one treatment group (red).
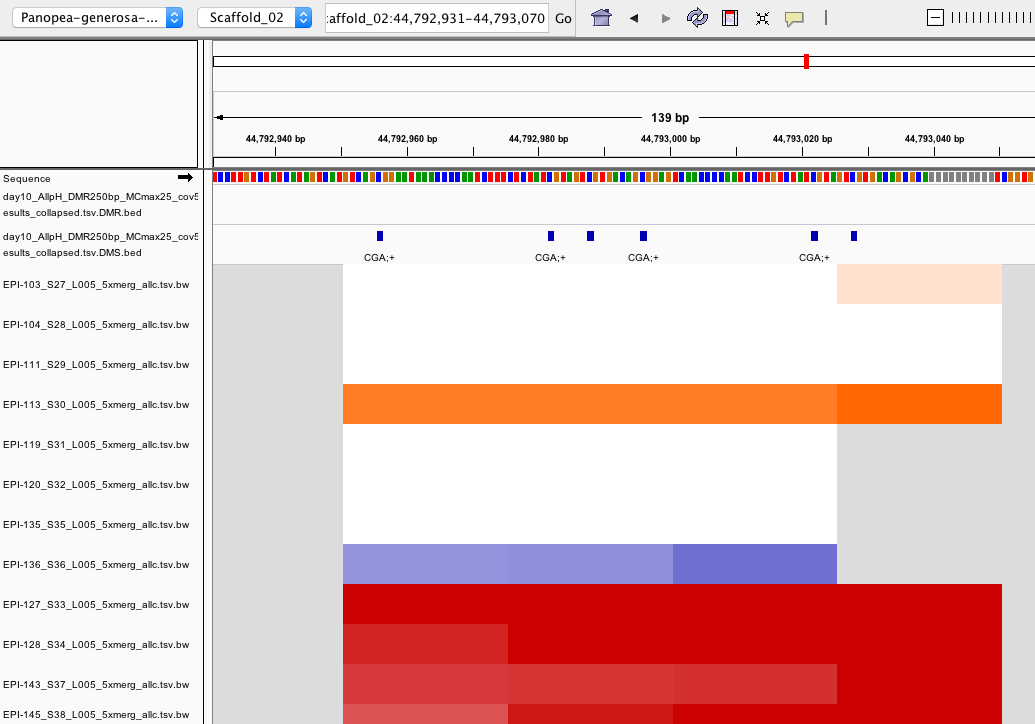
To me, this is showing that generally the blue and orange groups are not methylated at these DMS while the red group is. Why would DMRfind not classify these DMS as a DMR?
methylpy DMRfind \
--allc-files EPI-103_S27_L005_5xmerg_allc.tsv \
EPI-104_S28_L005_5xmerg_allc.tsv \
EPI-111_S29_L005_5xmerg_allc.tsv \
EPI-113_S30_L005_5xmerg_allc.tsv \
EPI-119_S31_L005_5xmerg_allc.tsv \
EPI-120_S32_L005_5xmerg_allc.tsv \
EPI-135_S35_L005_5xmerg_allc.tsv \
EPI-136_S36_L005_5xmerg_allc.tsv \
EPI-127_S33_L005_5xmerg_allc.tsv \
EPI-128_S34_L005_5xmerg_allc.tsv \
EPI-143_S37_L005_5xmerg_allc.tsv \
EPI-145_S38_L005_5xmerg_allc.tsv \
--samples EPI-103 EPI-104 EPI-111 EPI-113 EPI-119 EPI-120 EPI-135 EPI-136 EPI-127 EPI-128 EPI-143 EPI-145 \
--mc-type "CGN" \
--num-procs 8 \
--output-prefix day10_AllpH_DMR250bp_MCmax25_cov5x \
--mc-max-dist 25 \
--dmr-max-dist 250 \
--min-num-dms 3 \
--sample-category 0 0 1 1 2 2 2 2 0 0 1 1
Hi Yupeng,
I am very much interested in trying using Methylpy, but I am not familiar with Python and I am stuck with an error that I am not able to fix.
I am running this command:
$ methylpy paired-end-pipeline --read1-files FCH2N2NBBXY_L7_ARAsbmMAAAAAAA-9_1.fq.gz --read2-files FCH2N2NBBXY_L7_ARAsbmMAAAAAAA-9_2.fq.gz --sample L5_0mM_minimap2 --forward-ref L5_background_one_T-DNA_minimap_f.mmi --reverse-ref L5_background_one_T-DNA_minimap_r.mmi --ref-fasta L5_background_one_T-DNA.fa --compress-output T --num-procs 4 --trim-reads F --check-dependency T --generate-mpileup-file F --aligner minimap2
and get the following error (using either minimap2 or bowtie2 as an aligner):
Traceback (most recent call last):
File "/usr/local/bin/methylpy", line 5, in <module>
parse_args()
File "/Library/Python/2.7/site-packages/methylpy/parser.py", line 185, in parse_args
keep_temp_files=args.keep_temp_files)
File "/Library/Python/2.7/site-packages/methylpy/call_mc_pe.py", line 342, in run_methylation_pipeline_pe
keep_temp_files=keep_temp_files)
File "/Library/Python/2.7/site-packages/methylpy/call_mc_pe.py", line 1370, in call_methylated_sites_pe
keep_temp_files = keep_temp_files)
File "/Library/Python/2.7/site-packages/methylpy/call_mc_se.py", line 1498, in call_methylated_sites
+" -q "+str(min_mapq)+" -B -f "+reference_fasta+" "+inputf
TypeError: bad operand type for unary +: 'str'
Here is what I get when running the methylpy test:
Tue Mar 26 15:32:35 2019
Test importing methylpy module.
- importing call_mc_se: pass
- importing call_mc_pe: pass
- importing DMRfind: pass
- importing parser: pass
Check methylpy executable:pass
methylpy is successfully installed!
Check whether dependencies are available in PATH
- find bowtie: failed
- find bowtie2: found version 2.2.6
- find minimap2: found version 2.16-r922
- find samtools: found version 1.9
- find cutadapt: failed
- find java: found version 1.6.0_65
Only bowtie2 is available. Skip tests for bowtie related code
Test DMRfind: failed
Test reidentify-DMR: failed
Test build-reference with bowtie2: pass
Test build-reference with minimap2: pass
Test single-end-pipeline with bowtie2: pass
Test single-end-pipeline with minimap2: pass
Test paired-end-pipeline with bowtie2: pass
Test paired-end-pipeline with minimap2: pass
Test quality filter for BAM file of single-end data: pass
Test call-methylation-state for single-end data: pass
Test call-methylation-state for paired-end data: pass
Test merge-allc: pass
Test filter-allc: pass
All tests are done!
Tue Mar 26 15:34:09 2019
The tests took 94.28 seconds!
Many thanks for your help!
Hi,
I am attempting to perform the bowtie2 alignments within methylpy with reads allowed multiple mappings as in issue #19 . However, the --keep-temp-files True option does not seem to be keeping any temporary files around. I can see the initial .sam files being written during the run but then they are deleted by the end. I am not getting any errors, indicating that methylpy recognizes the flag and its argument.
For reference, my full command is
methylpy single-end-pipeline --read-files $READS --sample $NAME --forward-ref $FREF --reverse-ref $RREF --ref-fasta $FA_REF --num-procs 6 --keep-temp-files True --aligner-options \"-a\"
I am using methylpy 1.3.8 installed via pip within a python 3.7 conda environment.
Hi,
I get an error with methylpy with new picard version. Since picard has its own binary instead of jar file, methylpy is searching for picard.jar, can you please update this?
Here is the error
Error: Unable to access jarfile /picard.jar
Traceback (most recent call last):
File "~/.conda/envs/methylpy/bin/methylpy", line 5, in
parse_args()
File "~/.conda/envs/methylpy/lib/python3.7/site-packages/methylpy/parser.py", line 185, in parse_args
keep_temp_files=args.keep_temp_files)
File "~/.conda/envs/methylpy/lib/python3.7/site-packages/methylpy/call_mc_pe.py", line 276, in run_methylation_pipeline_pe
java_options=java_options)
File "~/.conda/envs/methylpy/lib/python3.7/site-packages/methylpy/call_mc_se.py", line 1338, in remove_clonal_bam
"QUIET=true"])
File "~/.conda/envs/methylpy/lib/python3.7/subprocess.py", line 328, in check_call
raise CalledProcessError(retcode, cmd)
subprocess.CalledProcessError: Command '['java', '-Djava.io.tmpdir=/scratch-cbe/tempFiles', '-jar', '/picard.jar', 'MarkDuplicates', 'INPUT=CBURWANXX_4#GCGTAGTA_719_libA_processed_reads.bam', 'OUTPUT=CBURWANXX_4#GCGTAGTA_719_libA_processed_reads_no_clonal.bam', 'ASSUME_SORTED=true', 'REMOVE_DUPLICATES=true', 'METRICS_FILE=CBURWANXX_4#GCGTAGTA_719_libA.metric', 'VALIDATION_STRINGENCY=LENIENT', 'QUIET=true']' returned non-zero exit status 1.
Cheers
r
Hi! yupenghe,
I am using methylpy first time and I am not very good in computer programming.
Any help will be much appreciated.
I am trying to map my paired-end reads to transposable elements of Arabidopsis with the following command line:
** methylpy paired-end-pipeline --read1-files New_Analysis/Data/Epiril368_4_C_R1_L001_R1.fastq.gz --read2-files New_Analysis/Data/Epiril368_4_C_R1_L001_R2.fastq.gz --sample New_Analysis/New_Output/TE/epiRIL368_4°C_Rep1_Lane1_TE --forward-ref New_Analysis/TE_Genome/TAIR10_TE_f --reverse-ref New_Analysis/TE_Genome/TAIR10_TE_r --ref-fasta New_Analysis/TE_Genome/TAIR10_TE.fas --generate-allc-file True --num-procs 4 --compress-output True --remove-clonal True --path-to-picard="picard/" **
But with alignment I got the following warnings:
Traceback (most recent call last):
File "/home/saurabh/anaconda3/bin/methylpy", line 4, in
import('pkg_resources').run_script('methylpy==1.3.4', 'methylpy')
File "/home/saurabh/anaconda3/lib/python3.6/site-packages/pkg_resources/init.py", line 664, in run_script
self.require(requires)[0].run_script(script_name, ns)
File "/home/saurabh/anaconda3/lib/python3.6/site-packages/pkg_resources/init.py", line 1444, in run_script
exec(code, namespace, namespace)
File "/home/saurabh/anaconda3/lib/python3.6/site-packages/methylpy-1.3.4-py3.6.egg/EGG-INFO/scripts/methylpy", line 5, in
parse_args()
File "/home/saurabh/anaconda3/lib/python3.6/site-packages/methylpy-1.3.4-py3.6.egg/methylpy/parser.py", line 185, in parse_args
keep_temp_files=args.keep_temp_files)
File "/home/saurabh/anaconda3/lib/python3.6/site-packages/methylpy-1.3.4-py3.6.egg/methylpy/call_mc_pe.py", line 342, in run_methylation_pipeline_pe
keep_temp_files=keep_temp_files)
File "/home/saurabh/anaconda3/lib/python3.6/site-packages/methylpy-1.3.4-py3.6.egg/methylpy/call_mc_pe.py", line 1343, in call_methylated_sites_pe
path_to_samtools=path_to_samtools)
File "/home/saurabh/anaconda3/lib/python3.6/site-packages/methylpy-1.3.4-py3.6.egg/methylpy/call_mc_pe.py", line 1265, in flip_read2_strand
for line in input_pipe.stdout:
File "/home/saurabh/anaconda3/lib/python3.6/codecs.py", line 321, in decode
(result, consumed) = self._buffer_decode(data, self.errors, final)
UnicodeDecodeError: 'utf-8' codec can't decode byte 0xb0 in position 509: invalid start byte
I don't understand exactly what it is.
Thank you!
Regards
CS791
Hi Yupeng, happy friday!
I am attempting to run DMRfind on some destranded bismark cytosine coverage files that I have reformatted to look like allc files. The files are located here.
Here is an example of the first 10 lines of one file:
Scaffold_01 209 + CGA 0 5 1
Scaffold_01 213 + CGA 0 5 1
Scaffold_01 221 + CGA 0 6 1
Scaffold_01 238 + CGA 0 8 1
Scaffold_01 565 + CGA 0 6 1
Scaffold_01 577 + CGA 0 6 1
Scaffold_01 583 + CGA 0 5 1
Scaffold_01 2509 + CGA 0 7 1
Scaffold_01 2554 + CGA 0 6 1
Scaffold_01 3002 + CGA 0 5 1
Because these are destranded I have included "+" for each cytosine loci and since the context was not included in the original file, I added "CGA" for each cytosine loci.
When I run DMRfind on these files, I get a weird error:
gsl: discrete.c:238: ERROR: probabilities must be non-negative
Default GSL error handler invoked.
gsl: discrete.c:238: ERROR: probabilities must be non-negative
Default GSL error handler invoked.
gsl: discrete.c:238: ERROR: probabilities must be non-negative
Default GSL error handler invoked.
Traceback (most recent call last):
File "/Users/strigg/anaconda3/bin/methylpy", line 5, in <module>
parse_args()
File "/Users/strigg/anaconda3/lib/python3.6/site-packages/methylpy/parser.py", line 71, in parse_args
seed=args.seed)
File "/Users/strigg/anaconda3/lib/python3.6/site-packages/methylpy/DMRfind.py", line 274, in DMRfind
convergence_diff=convergence_diff)
File "/Users/strigg/anaconda3/lib/python3.6/site-packages/methylpy/DMRfind.py", line 319, in merge_DMS_to_DMR
input_rms_file)
File "/Users/strigg/anaconda3/lib/python3.6/site-packages/methylpy/DMRfind.py", line 482, in get_resid_cutoff
residuals.append(float(resid))
ValueError: could not convert string to float: '-0.0Scaffold_01'
It seems like there is some strange formatting happening in the rms_results.tsv.gz file that causes the script to break and not produce the collapsed.tsv file.
I tried updating methylpy and gsl (and recompiling), but still got the same error. I ran the run_test.py script and everything passed, so I feel like it's probably my files. Here's my jupyter notebook documenting all this.
I also tried running this from another computer that methylpy has worked on and I get the same error (see my output logs here and here
Have you seen this behavior before? Any advice for moving forward? Thanks!
Hi, Yupeng,
Some suggestions of methylpy based on my experience. I can work on improve these (since I already did these here https://github.com/lhqing/cemba_data/blob/master/cemba_data/tools/). But I'd like to know your opinions:
chromosome names: I suggest always keep the chromosome name same as genome fasta, i.e. keep "chr" by default for genomes like mm10 and hg19, or never change chromosome names. I found this will decrease conflits in a lot cases.
chromosome orders: Right now the order of chrom in ALLC is based on bam file, and there is no paticular "sorted or not" check for ALLC file. What's worse is in the merge_allc_files_worker function, the order of chrom may be random, because dict.keys() is not ordered.
https://github.com/yupenghe/methylpy/blob/methylpy/methylpy/utilities.py#L543
Use bgzip/tabix:
More universal map_to_region function:
which is very important for single cell, my implementation here: map_to_region
Given an ALLC file, this function count mC and cov for a list of regions, e.g. chrom 100kb bins, gene bodies, I use bedtools map internally, which is very fast.
Hi, I found two minor bugs in call_mc.py although the bugs wouldn't affect the results much:
Line 2,746 in "call_methylated_sites"
avg_qual = total_phred / total_bases --> avg_qual = total_phred / float(total_bases)
Line 3,069 in "benjamini_hochberg_correction_call_methylated_sites"
bh_value = float(fields[6]) * mc_class_counts[fields[3]] / (test_num[fields[3] + 1) --> bh_value = float(fields[6]) * mc_class_counts[fields[3]] / test_num[fields[3]
When I run add-methylation-level with an input tsv (genes that are differentially expressed, for which I'm interested in getting total methylation level) methylpy generates a blank output file.
Hi Yupeng,
I am using methylpy to map 422 million paired-end reads (150 bp) to our draft genome (800 M bases), and get the "no space left on device" error. We reset TMPDIR to a directory with enough space (~4 T), but still get the same error. Could you please help us to figure out this problem? Please let me know if you need any other files.
Thanks!
Shan
Here is the script we used -- we use SLURM to submit our job:
#!/bin/bash
#SBATCH --job-name=Processing_Paired-end_Data_2.temp
#SBATCH --mail-user=[email protected]
#SBATCH --mail-type=FAIL,END
#SBATCH --output=Processing_Paired-end_Data_2.temp_%j.out
#SBATCH --error=Processing_Paired-end_Data_2.temp_%j.error
#SBATCH --nodes=1
#SBATCH --ntasks=1
#SBATCH --cpus-per-task=16
#SBATCH --mem-per-cpu=4gb
#SBATCH --time=30-00:00:00
date;hostname;pwd
export TMPDIR=/orange/soltis/shan158538/methylpy_temp
echo "Temp dir is: ${TMPDIR}"
module purge
module load gcc/5.2.0
module load methylpy/1.2.9
module load picard/2.18.3
methylpy paired-end-pipeline
--read1-files ${IN}/HMCWKCCXY_s8_1_4981-LF_17_SL334590.fastq.gz
--read2-files ${IN}/HMCWKCCXY_s8_2_4981-LF_17_SL334590.fastq.gz
--sample T.dubius_3060-1-4
--forward-ref ${DB}/Tdubius_Ref_f
--reverse-ref ${DB}/Tdubius_Ref_r
--ref-fasta ${DB}/Tdub_lambda.fasta
--path-to-output ${OUT}
--num-procs 16
--trim-reads True
--bowtie2 True
--remove-clonal True
--path-to-picard="$HPC_PICARD_DIR/"
--adapter-seq-read1 AGATCGGAAGAGCACACGTCTGAAC
--adapter-seq-read2 AGATCGGAAGAGCGTCGTGTAGGGA
--binom-test True
--unmethylated-control "NC_001416.1_lambda:"
--keep-temp-files True
Here are a few lines of the error message:
Exception in thread "main" htsjdk.samtools.util.RuntimeIOException: java.io.IOException: No space left on device
at htsjdk.samtools.util.SortingCollection.spillToDisk(SortingCollection.java:247)
at htsjdk.samtools.util.SortingCollection.doneAdding(SortingCollection.java:192)
at picard.sam.markduplicates.MarkDuplicates.buildSortedReadEndLists(MarkDuplicates.java:598)
at picard.sam.markduplicates.MarkDuplicates.doWork(MarkDuplicates.java:232)
at picard.cmdline.CommandLineProgram.instanceMain(CommandLineProgram.java:282)
at picard.cmdline.PicardCommandLine.instanceMain(PicardCommandLine.java:103)
at picard.cmdline.PicardCommandLine.main(PicardCommandLine.java:113)
Caused by: java.io.IOException: No space left on device
at sun.nio.ch.FileDispatcherImpl.write0(Native Method)
Here is our output:
Begin splitting reads for libA
Mon Jul 29 11:36:26 2019
Begin trimming reads for libA
Mon Jul 29 14:09:34 2019
Begin converting reads for libA
Mon Jul 29 14:27:14 2019
Begin Running Bowtie2 for libA
Mon Jul 29 14:37:34 2019
Processing forward strand hits
Tue Jul 30 05:06:11 2019
Processing reverse strand hits
Tue Jul 30 19:20:55 2019
Finding multimappers
Tue Jul 30 20:20:12 2019
Hi Yupeng!
I am looking for DMRs among 3 different categories with each category made up of 4 samples. I attempted to run DMRfind using the --min-cluster parameter set to 3 to require 3/4 samples per each category show coverage of the DMR. However, some DMRs returned had coverage of 3/4 samples from one category but only 1/4 samples from one other category and no coverage in any samples from the third category. For an example, see line 11 in the .txt (changed from .tsv for upload) results table created from the attached .sh.txt file. I interpreted category to mean treatment group. Am I misinterpreting what a category is or how this parameter should be used? Is there a away to require all groups or categories have coverage of a DMR in at least 3/4 samples per category?
Thanks,
Shelly
day135_AllpH_DMR250bp_MCmax25_cov5x_clst3_rms_results_collapsed.txt
Hi Yunpenghe,
I hope alls well with you.
I have a problem running DMRfind. which is giving me the below error. I think its not a file formate problem as I have been used the software well before and it might not be a dependencies problem because I run independently on the cluster and in anaconda environment and giving me the same error.
Could you please gimme a hit what might be happening.
Many Thanks,
Ranj
[ranjith.papareddy@clip-c2-2 Main_Cot_stem_aligned_bams]$ methylpy DMRfind --output-prefix CotvsStem.CHH --mc-type 'CHH' --min-cov 4 --num-sims 1000 --min-num-dms 4 --samples allc_LowerStem_F1_trimmed_bismark_bt2.Chsort allc_Cotyledon_C1_trimmed_bismark_bt2.Chsort --allc-files ../../methylation_man/Dev.series.DMRs/BC.WT.tsv ../../methylation_man/Dev.series.DMRs/Heart.WT.tsv
Filtering allc files using single node.
Wed Feb 5 14:30:52 2020
Splitting allc files for chromosome chloroplast
Wed Feb 5 14:32:24 2020
Running rms tests for chromosome chloroplast
Wed Feb 5 14:32:25 2020
/software/2020/software/methylpy/1.2.9-foss-2018b-python-2.7.15/lib/python2.7/site-packages/methylpy-1.2.9-py2.7.egg/methylpy/run_rms_tests.out: error while loading shared libraries: libgsl.so.0: cannot open shared object file: No such file or directory
(<class 'subprocess.CalledProcessError'>, 204)
Command '['/software/2020/software/methylpy/1.2.9-foss-2018b-python-2.7.15/lib/python2.7/site-packages/methylpy-1.2.9-py2.7.egg/methylpy/run_rms_tests.out', 'CotvsStem.CHH_filtered_allc_allc_LowerStem_F1_trimmed_bismark_bt2.Chsort.tsv_chloroplast_0.tsv,CotvsStem.CHH_filtered_allc_allc_Cotyledon_C1_trimmed_bismark_bt2.Chsort.tsv_chloroplast_0.tsv', 'CotvsStem.CHH_rms_results_for_chloroplast_chunk_0.tsv', 'allc_LowerStem_F1_trimmed_bismark_bt2.Chsort,allc_Cotyledon_C1_trimmed_bismark_bt2.Chsort', '4', '1000', '100', '-1']' returned non-zero exit status 127
Running RMS tests failed.
[ranjith.papareddy@clip-c2-2 Main_Cot_stem_aligned_bams]$ tail ../../methylation_man/Dev.series.DMRs/BC.WT.tsv
4 18585035 - CCT 18 20 1
4 18585036 - CCC 17 18 1
4 18585041 - CTA 15 15 1
4 18585042 - CCT 13 13 1
4 18585043 - CCC 10 10 1
4 18585048 - CTA 7 7 1
4 18585049 - CCT 7 7 1
4 18585050 - CCC 6 6 1
4 18585055 - CTA 1 1 1
4 18585056 - CCT 1 1 1
[ranjith.papareddy@clip-c2-2 Main_Cot_stem_aligned_bams]$
Hi,
I was trying to identify the DMRs using methylpy. But the results file is empty with just header.
The command and its output are here.
>methylpy DMRfind --allc-files allc_temp17.tsv.gz allc_temp18.tsv.gz allc_temp19.tsv.gz allc_temp20.tsv.gz --samples temp17 temp18 temp19 temp20 --mc-type CG --num-procs 8 --output-prefix run4
Splitting allc files for chromosome Chr1
Thu Dec 28 13:04:39 2017
Running rms tests for chromosome Chr1
Thu Dec 28 13:04:39 2017
Merging sorted run4_rms_results.tsv files.
Thu Dec 28 13:04:39 2017
Begin FDR Correction
Thu Dec 28 13:04:39 2017
m0 estimate for iteration 0: 0
Histogram FDR correction did not converge. Using the smallest p-value 0.000333333333333 as cutoff.
Calculating Residual Cutoff
Thu Dec 28 13:04:39 2017
There are no null residuals to calculate resid_cutoff. Using 2 as the cutoff.
Begin Defining Windows
Thu Dec 28 13:04:39 2017
Adding Methylation Levels
Thu Dec 28 13:04:39 2017
Done
Thu Dec 28 13:04:39 2017
Thanks,
Rahul
I ran methylpy for multiple samples, and then tried to reidentify DMRs in a subset of those samples. When I reidentify DMRs, I found some cases where the methylation levels do not correlate with the DMR labels. For instance, The methylation levels for a DMR in one sample is 0.78 and in the other is 0.68, but the latter is labeled as hypermethylated in the DMR reidentify results. What is the reason for this? I should note that compared to my remaining samples in the whole dataset, 0.68 is hypermethylated. But I don't understand why it still appears as hypermethylated when I am comparing it with just the 0.78 sample.
Could you also elaborate on the min_cluster option? I read the documentation and it's still not clear to me. Thanks!
Here are the parameters I used for initial DMR finding and DMR reidentify steps:
methylpy DMRfind --allc-files <A B C D E F G H> --mc-type "CGN" --sig-cutoff 0.01
methylpy reidentify-DMR --input-rms-file rms_results.tsv.gz --sig-cutoff 0.01 --min-num-dms 5 --collapse-samples A B
Hi Yupeng, I am trying to test my methylpy installation on a Linux machine, but the run_test.py fails at the stage of testing single-end-pipeline with bowtie. How can I fix this? Below is the terminal output:
$ python run_test.py
Tests start
Thu Aug 16 10:07:48 2018
Test importing methylpy module.
- importing call_mc_se: pass
- importing call_mc_pe: pass
- importing DMRfind: pass
- importing parser: pass
Check methylpy executable:pass
methylpy is successfully installed!
Check whether dependencies are available in PATH
- find bowtie: found version 1.2.2
- find bowtie2: found version 2.3.4.1
- find minimap2: failed
- find samtools: found version 1.9
- find cutadapt: found version 1.16
- find java: found version 1.8.0_171
Both bowtie and bowtie2 are available and will be tested.
Test DMRfind: pass
Test reidentify-DMR: pass
Test build-reference with bowtie: pass
Test build-reference with bowtie2: pass
Test single-end-pipeline with bowtie: Traceback (most recent call last):
File "run_test.py", line 204, in <module>
stderr=f_stderr)
File "/home/x/miniconda3/envs/methylpy/lib/python3.6/subprocess.py", line 291, in check_call
raise CalledProcessError(retcode, cmd)
subprocess.CalledProcessError: Command '['methylpy', 'single-end-pipeline', '--read-files', 'data/test_data_R1.fastq.gz', '--sample', 'se_bt', '--path-to-output', 'results/', '--forward-ref', 'chrL/chrL_f', '--reverse-ref', 'chrL/chrL_r', '--ref-fasta', 'data/chrL.fa', '--num-procs', '1', '--aligner', 'bowtie', '--bgzip', 'True', '--aligner', 'bowtie', '--path-to-aligner', '', '--path-to-samtools', '', '--remove-clonal', 'False', '--trim-reads', 'False', '--binom-test', 'True', '--unmethylated-control', '0.005']' returned non-zero exit status 1.
Here is the content of test_error_msg.txt generated :
/home/x/miniconda3/envs/methylpy/lib/python3.6/site-packages/scipy/stats/stats.py:1713: FutureWarning: Using a non-tuple sequence for multidimensional indexing is deprecated; use `arr[tuple(seq
)]` instead of `arr[seq]`. In the future this will be interpreted as an array index, `arr[np.array(seq)]`, which will result either in an error or a different result.
return np.add.reduce(sorted[indexer] * weights, axis=axis) / sumval
/home/x/miniconda3/envs/methylpy/lib/python3.6/site-packages/scipy/stats/stats.py:1713: FutureWarning: Using a non-tuple sequence for multidimensional indexing is deprecated; use `arr[tuple(seq
)]` instead of `arr[seq]`. In the future this will be interpreted as an array index, `arr[np.array(seq)]`, which will result either in an error or a different result.
return np.add.reduce(sorted[indexer] * weights, axis=axis) / sumval
Building a SMALL index
Building a SMALL index
# reads processed: 93996
# reads with at least one reported alignment: 44539 (47.38%)
# reads that failed to align: 49457 (52.62%)
Reported 44539 alignments
# reads processed: 93996
# reads with at least one reported alignment: 44443 (47.28%)
# reads that failed to align: 49553 (52.72%)
Reported 44443 alignments
[mpileup] 1 samples in 1 input files
Traceback (most recent call last):
File "/home/x/miniconda3/envs/methylpy/bin/methylpy", line 4, in <module>
__import__('pkg_resources').run_script('methylpy==1.3.4', 'methylpy')
File "/home/x/miniconda3/lib/python3.6/site-packages/pkg_resources/__init__.py", line 654, in run_script
self.require(requires)[0].run_script(script_name, ns)
File "/home/x/miniconda3/lib/python3.6/site-packages/pkg_resources/__init__.py", line 1434, in run_script
exec(code, namespace, namespace)
File "/shares/x_share/private/x/miniconda3/envs/methylpy/lib/python3.6/site-packages/methylpy-1.3.4-py3.6.egg/EGG-INFO/scripts/methylpy", line 5, in <module>
parse_args()
File "/home/x/miniconda3/envs/methylpy/lib/python3.6/site-packages/methylpy-1.3.4-py3.6.egg/methylpy/parser.py", line 134, in parse_args
keep_temp_files=args.keep_temp_files)
File "/home/x/miniconda3/envs/methylpy/lib/python3.6/site-packages/methylpy-1.3.4-py3.6.egg/methylpy/call_mc_se.py", line 323, in run_methylation_pipeline
keep_temp_files=keep_temp_files)
File "/home/x/miniconda3/envs/methylpy/lib/python3.6/site-packages/methylpy-1.3.4-py3.6.egg/methylpy/call_mc_se.py", line 1644, in call_methylated_sites
bgzip_allc_file(output_file,path_to_bgzip,path_to_tabix,buffer_line_number)
File "/home/x/miniconda3/envs/methylpy/lib/python3.6/site-packages/methylpy-1.3.4-py3.6.egg/methylpy/utilities.py", line 66, in bgzip_allc_file
output_pipe.stdin.write(out)
TypeError: a bytes-like object is required, not 'str'
the allc file has all 1's even when the coverage is bad.
Ex.
1 2 + CCT 1 3 1
1 3 + CTA 1 1 1
1 8 + CCC 1 1 1
1 9 + CCT 1 1 1
1 10 + CTA 1 1 1
1 15 + CCC 1 1 1
1 16 + CCT 1 1 1
1 17 + CTA 1 1 1
1 22 + CCC 1 2 1
1 23 + CCT 1 2 1
1 24 + CTA 2 2 1
1 29 + CCT 0 2 1
1 30 + CTC 0 2 1
1 32 + CTG 0 2 1
Hi ,
With methylpy paired or single end pipe line, if i havent missed, are there any input options for methylpy to trim 5' base in extract with normal adaptor trimming to to mitigate Mbias.
Many Thanks,
Ranj
Hi Yupeng,
Sorry for bothering yet again. I noticed that PBAT alignment mode is not implemented in PairedEnd mode(at-least thats what its said in call_mc_pe.py). Is it implemented in SingleEnd mode? I didn't find any apparent change with alignment with PBAT option set to True or False.
Many thanks & have a good one.
Ranj
Dear Yupeng,
Methylpy output showed a low rate of uniquely mapping read pairs:
There are 258651069 total input read pairs
Wed Dec 25 08:40:05 2019
There are 15891937 uniquely mapping read pairs, 6.14416057179 percent remaining
Wed Dec 25 08:40:05 2019
There are 13360495 non-clonal read pairs, 5.16545129763 percent remaining
Wed Dec 25 08:40:05 2019
For the same dataset, however, the bowtie2 mapping report showed a pretty high mapping rate:
258648925 reads; of these:
258648925 (100.00%) were paired; of these:
109280474 (42.25%) aligned concordantly 0 times
26355304 (10.19%) aligned concordantly exactly 1 time
123013147 (47.56%) aligned concordantly >1 times
57.75% overall alignment rate
258648925 reads; of these:
258648925 (100.00%) were paired; of these:
109452788 (42.32%) aligned concordantly 0 times
26285006 (10.16%) aligned concordantly exactly 1 time
122911131 (47.52%) aligned concordantly >1 times
57.68% overall alignment rate
I am curious about why would this happen. Could you please take a look? Does this dataset look bad? Please let me know if you need any other information.
Thanks and happy holidays!!
Shan
Hi, I have similar issue as #12 , but I can't solve it.
When I qsub a job for methylpy allc-to-bigwig with 1 or 10 CPU core and 10G memory, it sometimes shows the error and end the process:
Command:
methylpy allc-to-bigwig --allc-file allc_hs_NeuN_neg.tsv --output-file allc_NeuN_neg_mCG_bin1.bw --ref-fasta hg19/genome/fasta/hg19.fa --mc-type CGN --bin-size 1
Traceback (most recent call last): File "anaconda3/envs/syniu/bin/methylpy", line 4, in <module> __import__('pkg_resources').run_script('methylpy==1.3.4', 'methylpy') File "anaconda3/envs/syniu/lib/python3.6/site-packages/pkg_resources/__init__.py", line 664, in run_script self.require(requires)[0].run_script(script_name, ns) File "anaconda3/envs/syniu/lib/python3.6/site-packages/pkg_resources/__init__.py", line 1444, in run_script exec(code, namespace, namespace) File "anaconda3/envs/syniu/lib/python3.6/site-packages/methylpy-1.3.4-py3.6.egg/EGG-INFO/scripts/methylpy", line 5, in <module> parse_args() File "anaconda3/envs/syniu/lib/python3.6/site-packages/methylpy-1.3.4-py3.6.egg/methylpy/parser.py", line 327, in parse_args add_chr_prefix=args.add_chr_prefix) File "anaconda3/envs/syniu/lib/python3.6/site-packages/methylpy-1.3.4-py3.6.egg/methylpy/utilities.py", line 252, in convert_allc_to_bigwig +output_file),stderr=subprocess.PIPE) File "anaconda3/envs/syniu/lib/python3.6/subprocess.py", line 291, in check_call raise CalledProcessError(retcode, cmd) subprocess.CalledProcessError: Command '['wigToBigWig', 'mCH_bin1.bw.wig', 'mCH_bin1.bw.chrom_size', 'mCH_bin1.bw']' returned non-zero exit status 1.
First few line of chrom.size file
chr1 249250621 chr2 243199373 chr3 198022430 chr4 191154276 chr5 180915260 chr6 171115067 chr7 159138663 chrX 155270560 chr8 146364022 chr9 141213431 chr10 135534747 chr11 135006516 chr12 133851895 chr13 115169878 chr14 107349540 chr15 102531392 chr16 90354753 chr17 81195210 chr18 78077248 chr20 63025520 chrY 59373566 chr19 59128983 chr22 51304566 chr21 48129895
few lines of wig file:
chr1 11992 11993 0.1 chr1 11994 11995 0.0 chr1 11995 11996 0.0 chr1 11997 11998 0.0 chr1 12000 12001 0.15384615384615385 chr1 12003 12004 0.0 chr1 12005 12006 0.0 chr1 12006 12007 0.1
chromosome name in .fa file is like chr1
It works sometimes when I restart it, but sometimes it doesn't. I can see the wigToBigWig help page when i use it in terminal. I also use around 10 CPU. Not sure if it's the subprocess issue.
Do you have any suggestion?
Hello,
I am new at BS-seq experiments ansd was hoping to get some help with the problem i encounter.
I have paired-end illumina fastq files (~350M reads R1 and ~350m reads R2) that were trimmed and clipped to remove adapters (with trim_galore) and then fastqc checked.
I am now trying to run methylpy 1.3.0 to align and extract methylation data.
The command I run is as follow:
methylpy paired-end-pipeline
--read1-files fileR1.fq.gz
--read2-files fileR2.fq.gz
--sample sample name
--forward-ref /path to/ genome_mm10_f
--reverse-ref /path to/ genome_mm10_r
--ref-fasta /path/to/mm10.fa
--path-to-output \path/to/output
--trim-reads False
--check-dependency True
--generate-allc-file True
--generate-mpileup-file True
--compress-output True
--aligner bowtie2
--path-to-aligner /path/to/bowtie2/2.3.2/
--aligner-options ["--dovetail","--fr"]
--remove-clonal True
--path-to-picard /share/pkg/picard/2.17.8
--path-to-samtools /path/to/samtools/1.4.1/bin
--binom-test True \
As an output I get:
two fastq files named:
file_name_libA_converted_reads1.fastq
file_name_libA_converted_reads2.fastq
one sam file:
file_name_libA_forward_strand_hits.sam
my problem is:
the process never goes past that step
the sam file is empty
I also get a warning message:
Warning: Output file '[--dovetail,--fr,-S]' was specified without -S. This will not work in future Bowtie 2 versions. Please use -S instead.
I have tried to pass the -S option to Bowtie2 but still get the same warning.
As anybody encounterred the same problem?
I wonder if the alignment parameters are off in some way but I can't figure out what to change.
Any help would be greatly appreciated
Thanks!
Hello,
I have been trying to install methylpy, but when I run the test methylpy, I get the attached error messages. I am installing on ubuntu. Any information you could provide to fix the problem would be much appreciated.
Thank you
test_error_msg.txt
test_output_msg.txt
While running DMRFind module I get this warning:
/home/x/miniconda3/envs/methylpy/lib/python3.6/site-packages/scipy/stats/stats.py:1713: FutureWarning: Using a non-tuple sequence for multidimensional indexing is deprecated; use 'arr[tuple(seq)]' instead of 'arr[seq]'. In the future this will be interpreted as an array index, 'arr[np.array(seq)]', which will result either in an error or a different result. return np.add.reduce(sorted[indexer] * weights, axis=axis) / sumval
I am assuming this won't be a problem for my current analysis?
I have a similar issue to #48
But my output tsv file has only the bed file
This is the test bed file
chromosome start end
2 1 40001
2 40001 80001
2 80001 120001
2 120001 160001
This is the output I get
chromosome start end methylation_level_ACA methylation_level_ACB methylation_level_pCMT3-RNAiA methylation_level_pCMT3-RNAiB
2 1 40001
2 40001 80001
2 80001 120001
2 120001 160001
I checked that my files are using proper tab as delimiter and the allc files are not empty....
2 1647 - CAT 2 20 1
2 1649 + CGT 10 12 1
2 1650 - CGT 16 21 1
2 1653 + CCT 0 12 0
Here is the code
methylpy add-methylation-level
--input-tsv-file methylpy_wgenome.tsv
--output-file $outfile
--allc-files allc_ACA.tsv.gz allc_ACB.tsv.gz allc_pCMT3-RNAiA.tsv.gz allc_pCMT3-RNAiB.tsv.gz
--samples ACA ACB pCMT3-RNAiA pCMT3-RNAiB
--mc-type CHH
--num-procs 10
======
update- this is interesting. In the end I figured that it is because the allc files are not sorted. Shouldn't this be sorted automatically? This is my pair end pipeline code, did I miss anything???
methylpy paired-end-pipeline
--read1-files ./_1.fq.gz --read2-files ./_2.fq.gz --sample ${folderall[$indi]}
--merge-by-max-mapq True
--binom-test True
--unmethylated-control chloroplast
--min-cov 3
--forward-ref $fref --reverse-ref $rref --ref-fasta $reff
--num-procs 20 --sort-mem 35000000000
--path-to-output $directout
--remove-clonal True
--path-to-picard="/home/softwares/picard/"
--aligner-options "-p 8"
--trim-read False
1>$stdoutfile 2>$errfile
Hello,
I have been getting ValueErrors when I attempt to map samples using methylpy 1.3.2 during the sorting steps after SAM file creation.
I have attached two examples of the error file output. Any help in how I can resolve this error would be most appreciated.
error_methylpy2.txt
error_methylpy.txt
Thank you!
A declarative, efficient, and flexible JavaScript library for building user interfaces.
🖖 Vue.js is a progressive, incrementally-adoptable JavaScript framework for building UI on the web.
TypeScript is a superset of JavaScript that compiles to clean JavaScript output.
An Open Source Machine Learning Framework for Everyone
The Web framework for perfectionists with deadlines.
A PHP framework for web artisans
Bring data to life with SVG, Canvas and HTML. 📊📈🎉
JavaScript (JS) is a lightweight interpreted programming language with first-class functions.
Some thing interesting about web. New door for the world.
A server is a program made to process requests and deliver data to clients.
Machine learning is a way of modeling and interpreting data that allows a piece of software to respond intelligently.
Some thing interesting about visualization, use data art
Some thing interesting about game, make everyone happy.
We are working to build community through open source technology. NB: members must have two-factor auth.
Open source projects and samples from Microsoft.
Google ❤️ Open Source for everyone.
Alibaba Open Source for everyone
Data-Driven Documents codes.
China tencent open source team.