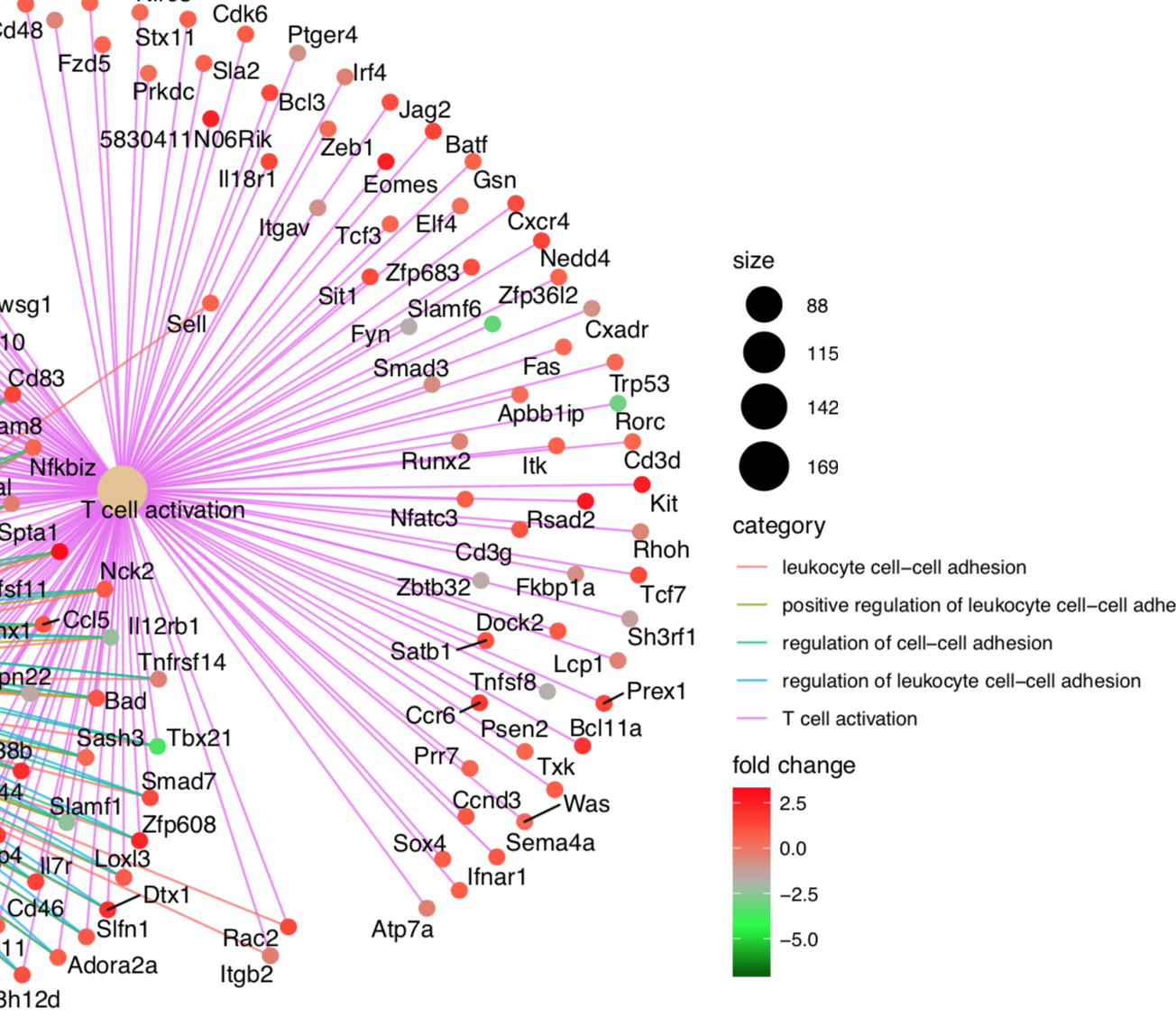
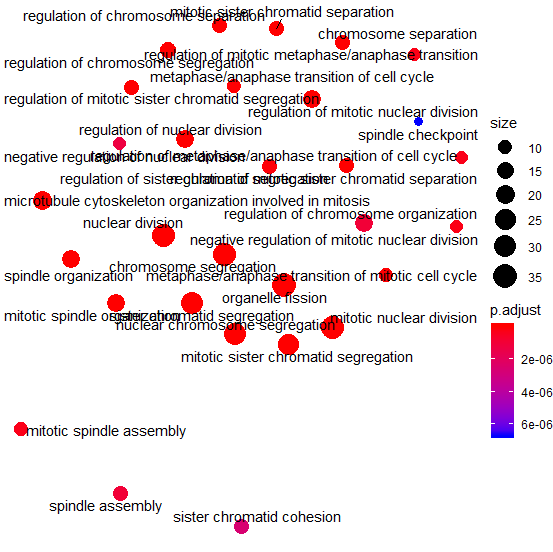
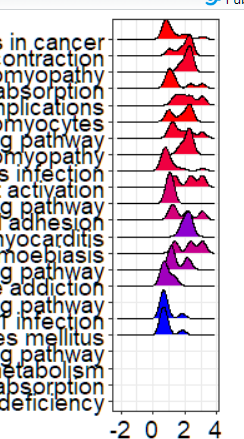
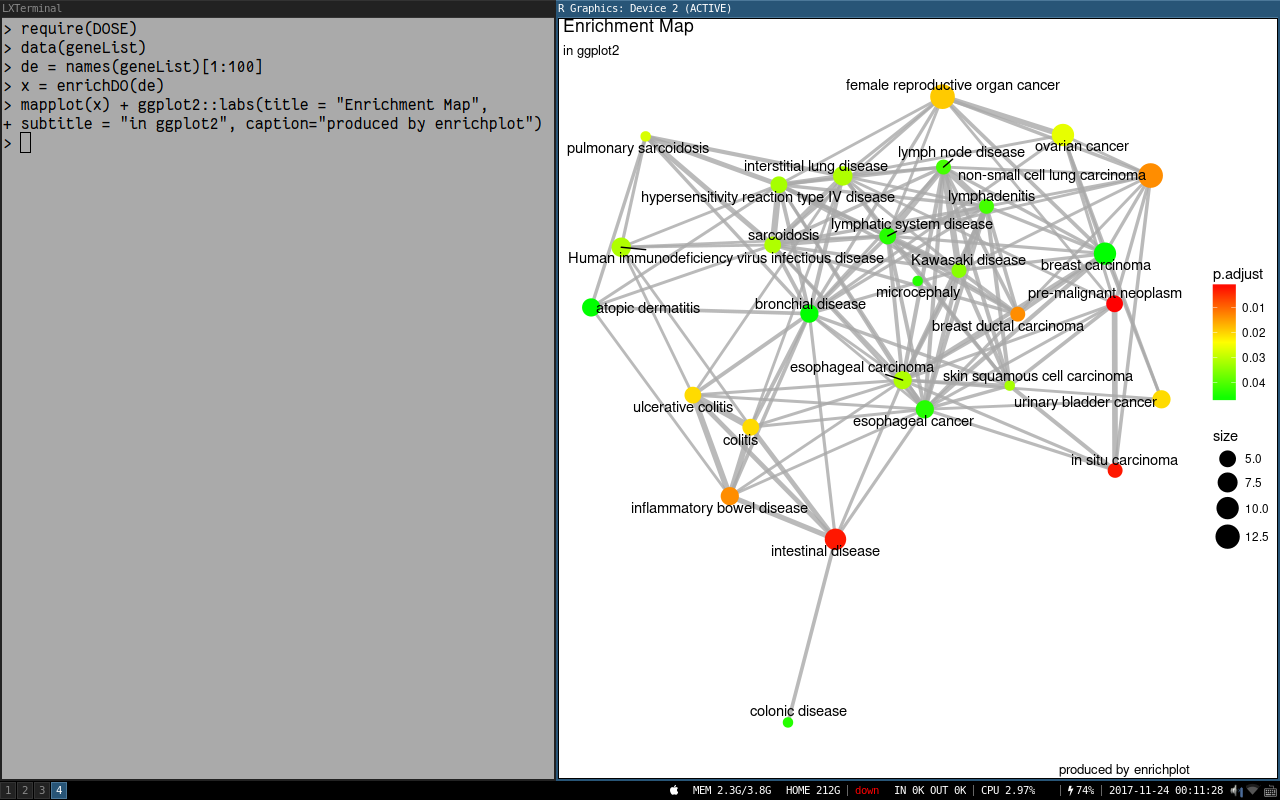
Hello! This has only been occurring recently -- but dotplot throws a strange warning when applied to objects of 'gseaResult' ---
orderBy appears to be an internal parameter, so it's a bit confusing to get this error when it doesn't appear that there was an obvious way to set 'orderBy' in the first place.
Anyways -- also wanted to say I love the clusterProfiler/enrichPlot suite of tools, thank you for all the work your team does!! Hope this helps!
# Wrangle count data
counts <- read.table("Data/Quiescent.vs.Proliferative.Raw.Counts.txt", header = T, row.names = 1)
colData <- data.frame(sample = colnames(counts), condition = c(rep("Quiescent", 3), rep("Proliferative", 3)))
colData$condition <- factor(colData$condition, levels = c("Quiescent", "Proliferative"))
dds <- DESeqDataSetFromMatrix(countData = counts, colData = colData, design = ~condition)
# QC
keep <- rowSums(counts(dds)) >= 10
dds <- dds[keep,]
# DGE
dds <- DESeq(dds)
res <- results(dds, contrast = c("condition", "Quiescent", "Proliferative"))
resDF <- as.data.frame(res)
resDF$geneID <- rownames(resDF)
# wrange DGE results
ensembl <- useMart("ensembl",dataset="hsapiens_gene_ensembl", host = "useast.ensembl.org")
map <- getBM(attributes = c("ensembl_gene_id", "description", "external_gene_name"), mart = ensembl)
colnames(map) <- c("geneID", "description", "geneName")
resDF <- merge(x = map, y = resDF, by = "geneID")
resDF <- resDF[,c(3,2,1,4:9)]
resDF$GSEA <- -log10(resDF$padj) * sign(resDF$log2FoldChange)
resDF <- resDF[order(resDF$GSEA, decreasing = T),]
# GSEA
geneList <- resDF$GSEA
names(geneList) <- resDF$geneName
geneList <- geneList[which(! duplicated(names(geneList)))]
geneList <- geneList[which(! is.na(geneList))]
TERM2GENE <- msigdbr()
TERM2GENE <- TERM2GENE[,c(1,5)]
EGMT <- GSEA(geneList = geneList, TERM2GENE = TERM2GENE, pvalueCutoff = .2)
enrichplot::dotplot(EGMT)`
R version 3.6.0 (2019-04-26)
Platform: x86_64-w64-mingw32/x64 (64-bit)
Running under: Windows 10 x64 (build 17763)
Matrix products: default
locale:
[1] LC_COLLATE=English_United States.1252 LC_CTYPE=English_United States.1252 LC_MONETARY=English_United States.1252
[4] LC_NUMERIC=C LC_TIME=English_United States.1252
attached base packages:
[1] parallel stats4 stats graphics grDevices utils datasets methods base
other attached packages:
[1] pheatmap_1.0.12 biomaRt_2.40.0 msigdbr_6.2.1 tibble_2.1.1
[5] dplyr_0.8.1 EnhancedVolcano_1.2.0 ggrepel_0.8.1 ggplot2_3.1.1
[9] clusterProfiler_3.12.0 DESeq2_1.24.0 SummarizedExperiment_1.14.0 DelayedArray_0.10.0
[13] BiocParallel_1.18.0 matrixStats_0.54.0 Biobase_2.44.0 GenomicRanges_1.36.0
[17] GenomeInfoDb_1.20.0 IRanges_2.18.0 S4Vectors_0.22.0 BiocGenerics_0.30.0
loaded via a namespace (and not attached):
[1] fgsea_1.10.0 colorspace_1.4-1 ggridges_0.5.1 qvalue_2.16.0 htmlTable_1.13.1
[6] XVector_0.24.0 base64enc_0.1-3 rstudioapi_0.10 farver_1.1.0 urltools_1.7.3
[11] bit64_0.9-7 AnnotationDbi_1.46.0 xml2_1.2.0 splines_3.6.0 GOSemSim_2.10.0
[16] geneplotter_1.62.0 knitr_1.23 polyclip_1.10-0 Formula_1.2-3 jsonlite_1.6
[21] SMVar_1.3.3 packrat_0.5.0 annotate_1.62.0 cluster_2.0.8 GO.db_3.8.2
[26] ggforce_0.2.2 BiocManager_1.30.4 compiler_3.6.0 httr_1.4.0 rvcheck_0.1.3
[31] backports_1.1.4 assertthat_0.2.1 Matrix_1.2-17 lazyeval_0.2.2 limma_3.40.2
[36] tweenr_1.0.1 acepack_1.4.1 htmltools_0.3.6 prettyunits_1.0.2 tools_3.6.0
[41] igraph_1.2.4.1 gtable_0.3.0 glue_1.3.1 GenomeInfoDbData_1.2.1 reshape2_1.4.3
[46] DO.db_2.9 fastmatch_1.1-0 Rcpp_1.0.1 enrichplot_1.4.0 gdata_2.18.0
[51] ggraph_1.0.2 xfun_0.7 stringr_1.4.0 gtools_3.8.1 XML_3.98-1.19
[56] DOSE_3.10.0 europepmc_0.3 zlibbioc_1.30.0 MASS_7.3-51.4 scales_1.0.0
[61] hms_0.4.2 RColorBrewer_1.1-2 curl_3.3 memoise_1.1.0 gridExtra_2.3
[66] UpSetR_1.4.0 triebeard_0.3.0 rpart_4.1-15 metaMA_3.1.2 latticeExtra_0.6-28
[71] stringi_1.4.3 RSQLite_2.1.1 genefilter_1.66.0 checkmate_1.9.3 caTools_1.17.1.2
[76] rlang_0.3.4 pkgconfig_2.0.2 bitops_1.0-6 lattice_0.20-38 purrr_0.3.2
[81] labeling_0.3 htmlwidgets_1.3 cowplot_0.9.4 bit_1.1-14 tidyselect_0.2.5
[86] plyr_1.8.4 magrittr_1.5 R6_2.4.0 gplots_3.0.1.1 Hmisc_4.2-0
[91] DBI_1.0.0 withr_2.1.2 pillar_1.4.0 foreign_0.8-71 survival_2.44-1.1
[96] RCurl_1.95-4.12 nnet_7.3-12 crayon_1.3.4 KernSmooth_2.23-15 viridis_0.5.1
[101] progress_1.2.2 locfit_1.5-9.1 grid_3.6.0 data.table_1.12.2 blob_1.1.1
[106] digest_0.6.19 xtable_1.8-4 tidyr_0.8.3 gridGraphics_0.4-1 munsell_0.5.0
[111] viridisLite_0.3.0 ggplotify_0.0.3