Interpretation of RNA-seq experiments through robust, efficient comparison to public databases
Thousands of RNA sequencing profiles have been deposited in public archives, yet remain unused for the interpretation of most newly performed experiments. Methods for leveraging these public resources have focused on the interpretation of existing data, or analysis of new datasets independently, but do not facilitate direct comparison of new to existing experiments. The interpretability of common unsupervised analysis methods such as Principal Component Analysis would be enhanced by efficient comparison of the results to previously published datasets.
To help identify replicable and interpretable axes of variation in any given gene expression dataset, we performed principal component analysis (PCA) on 536 studies comprising 44,890 RNA sequencing profiles. Sufficiently similar loading vectors, when compared across studies, were combined through simple averaging. We annotated the collection of resulting average loading vectors, which we call Replicable Axes of Variation (RAV), with details from the originating studies and gene set enrichment analysis. Functions to match PCA of new datasets to RAVs from existing studies, extract interpretable annotations, and provide intuitive visualization, are implemented as the GenomicSuperSignature R package, to be submitted to Bioconductor.
Usecases and benchmark examples are documented in the GenomicSuperSignaturePaper page. All the figures and tables can be reproduced using the code and instruction in this page as well.
If you use GenomicSuperSignature in published research, please cite:
Oh S, Geistlinger L, Ramos M, Blankenberg D, van den Beek M, Taroni JN, Carey VJ, Waldron L, Davis S. GenomicSuperSignature facilitates interpretation of RNA-seq experiments through robust, efficient comparison to public databases. Nature Communications 2022;13: 3695. doi: 10.1038/s41467-022-31411-3
The workflow to build the RAVmodel is available from https://github.com/shbrief/model_building which is archived in Zenodo with the identifier https://doi.org/10.5281/zenodo.6496552. All analyses presented in the GenomicSuperSignatures manuscript are reproducible using code accessible from https://github.com/shbrief/GenomicSuperSignaturePaper/ and archived in Zenodo with the identifier https://doi.org/10.5281/zenodo.6496612.
You can install GenomicSuperSignature in Bioconductor. This can be done using
BiocManager:
if (!require("BiocManager"))
install.packages("BiocManager")
library(BiocManager)
install("GenomicSuperSignature")
RAVmodel can be directly downloaded from Google bucket with no cost. The sizes of
RAVmodelsRAVmodel_C2.rds and RAVmodel_PLIERpriors.rds are 476.1MB and 475.1MB,
respectively. You can use wget or GenomicSuperSignature::getModel function.
## Download RAVmodel with wget
wget https://storage.googleapis.com/genomic_super_signature/RAVmodel_C2.rds
wget https://storage.googleapis.com/genomic_super_signature/RAVmodel_PLIERpriors.rds
## Download RAVmodel with getModel function
getModel("C2")
getModel("PLIERpriors")
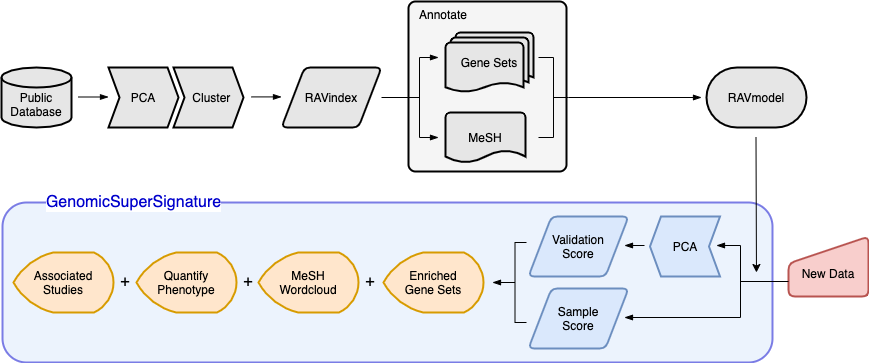
Schematic illustration of RAVmodel construction and GenomicSuperSignature application. Building the RAVmodel (components in grey) is performed once on a time scale of hours on a high-memory, high-storage server. Users can apply RAVmodel on their data (component in red) using the GenomicSuperSignature R/Bioconductor package (components in blue), which operates on a time scale of seconds for exploratory data analyses (components in orange) on a typical laptop computer.
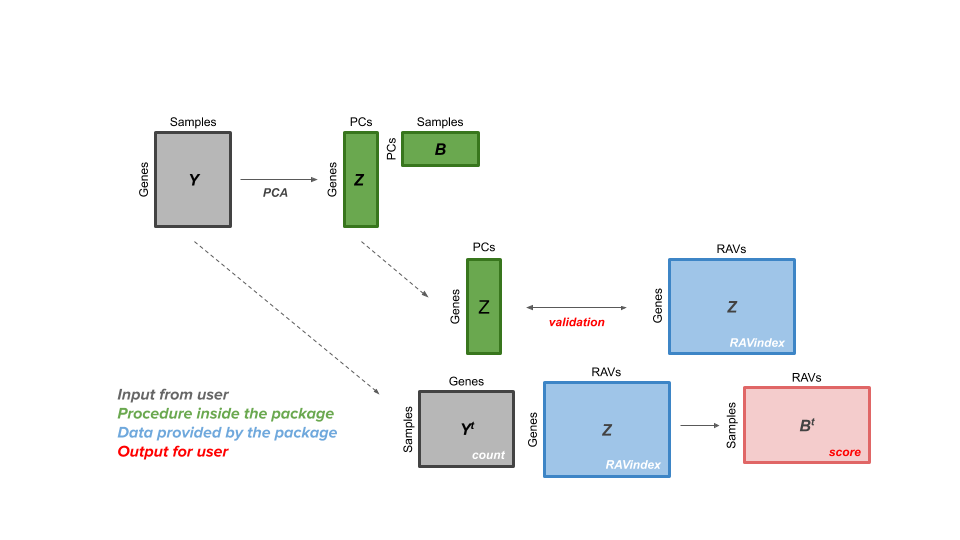
The GenomicSuperSignature package allows users to access a RAVmodel (Z matrix, blue) and annotation information on each RAV. From a gene expression matrix (Y matrix, grey), users can calculate dataset-level validation score or sample score matrix (B matrix, red). Through the RAV of your interest, additional information such as related studies, GSEA, and MeSH terms can be easily extracted.
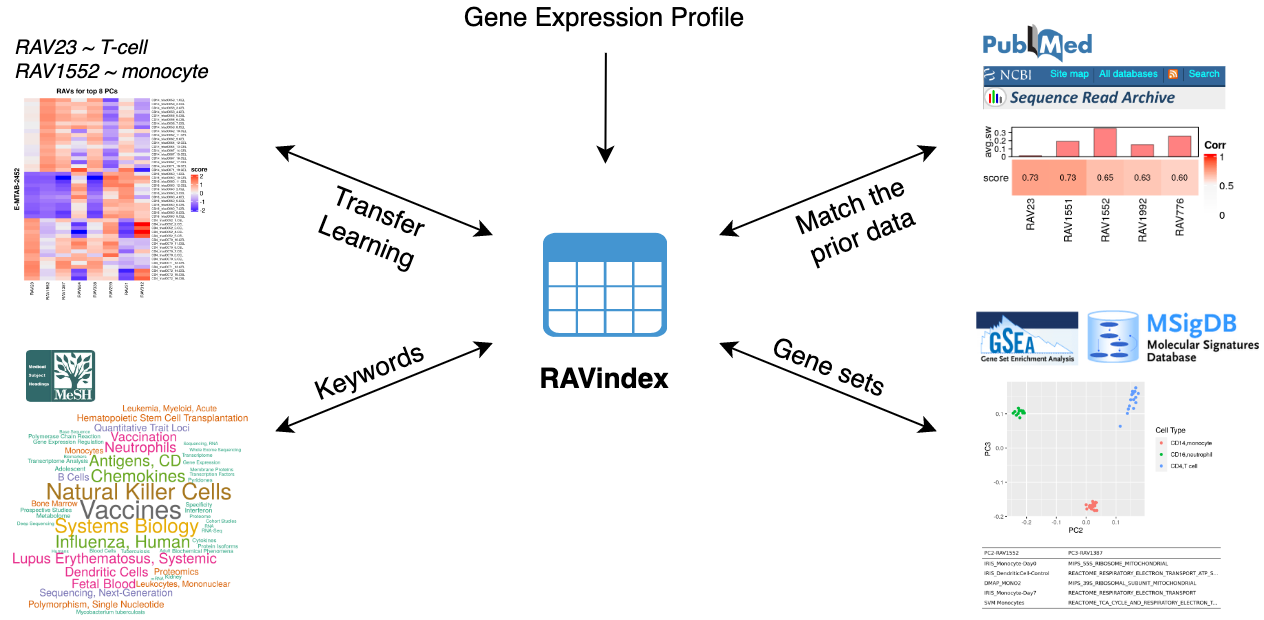
GenomicSuperSignature connects different public databases and prior information through RAVindex, creating the knowledge graph illustrated here. Users can instantly access data and metadata resources from multiple entry points, such as gene expression profiles, MeSH terms, gene sets, and keywords.