mannlabs / alphapept Goto Github PK
View Code? Open in Web Editor NEWA modular, python-based framework for mass spectrometry. Powered by nbdev.
Home Page: https://mannlabs.github.io/alphapept/
License: Apache License 2.0
A modular, python-based framework for mass spectrometry. Powered by nbdev.
Home Page: https://mannlabs.github.io/alphapept/
License: Apache License 2.0
Describe the bug
Currently people can select 'lfq_quantification' without ever having imported or searched any file. It would be important to make the selection of workflow steps somehow linked to whether an hdf file with required information is already there.
Importantly, in these cases, the settings files need to somehow be saved differently to ensure that users can still follow how the final results were created in case multiple different settings were used and then stacked on top of each other.
Greetings,
While reviewing the AlphaPept code, I noticed that the io.py module has a number of functions which appear to be repeated, as noted below.
Describe the bug
The Win GUI crashes and reports "PermissionError: [ WinError 5]".
To Reproduce
Steps to reproduce the behavior:
Expected behavior
Expected analysis to start
Screenshots

Version (please complete the following information):
Additional context
After receiving the error I tried to run as admin, but I was unable to load any raw files or fast files to the GUI.
Describe the bug
A list of paths to fasta files is always empty in the "Settings" tab in GUI. It can't be modified and not updated with the paths to fasta files that are selected in the "Experiment" tab.
To Reproduce
Screenshots
https://i.gyazo.com/b7962a7232e288fd1a65b003f9101b7c.png
Version (please complete the following information):
Deopendabot currently pushedsto master. This means that develop is behind master, which should never happen normally. Perhaps it is best to update dependabot to push to develop, so that master is always the "correct release version"?
Describe the bug
[Medium] "UnboundLocalError: local variable 'file' referenced before assignment" error in the Results section when you start the AP for the first time or when user cleans the "alphapept/finished" folder.
To Reproduce
Expected behavior
It would be nice to avoid showing the error when the folder is still empty.
Screenshots
https://gyazo.com/5d207434bd44156650f1612f8c1fe9e8
Version (please complete the following information):
Is your feature request related to a problem? Please describe.
I find it a bit unintuitive that you first select workflow steps, but then you can still choose settings for parts of the workflow that you didn't select.

Here, matching is not selected in the workflow, but I can still adjust parameters for it.
Describe the solution you'd like
Would it be possible to restrict the settings to the ones that are actually relevant for the workflow steps that were chosen?
Describe the solution you'd like
Now the tab with the tool calls ‘webui – Streamlit’. It would be nice to change it to AlphaPept with the proper logo if it’s possible.
@jalew188 already encountered the following issue, which I can now confirm: Line endings of Jupter notebooks on Window include a carriage return, which unfortunately means that a nbdev_build_lib or nbdev_clean_nbs function creates massive "differences", which are in fact just line endings. This obscures which code has been modified though, making it difficult to work on both windows and macOS/linux machine simultaneously...
Is your feature request related to a problem? Please describe.
If a user makes a check of the settings specified for the run in the "Settings" tab and no problem were found he gets the following message:
"Found a total of 0 problems: []" (see the screenshot - https://i.gyazo.com/8a515079cffed3dba4f92ffdce1123f8.png)
Describe the solution you'd like
It would be better to change the message for the case when no problems were found in the result of the check to something like:
"No problems were found."
The GUI seems extremely slow compared to the CLI (thermo quick test approx 4min instead of < 2min) on a. MacBook. Am I the first/only to notice this, or is this is a common issue/bug?
Describe the bug
With h5py==3.2.1, pandas raised an error: ERROR:root:Scoring of file d:\DataSets\APTest\20170518_QEp1_FlMe_SA_BOX0_HeLa12_Ecoli1_Shotgun.ms_data.hdf failed. Exception Can only use .str accessor with string values!. alphapept could run smoothly after switching to h5py==2.10.0. I check the data frame from .ms_data.hdf, it seems that strings are loaded (or stored) as binary strings from HDF_File in h5py==3.2.1.
To Reproduce
Install h5py==3.2.1, and run the whole workflow.
Expected behavior
Error ...... Can only use .str accessor with string values!
Version (master branch, 3.11-dev0):
I used alphapept import to extract ms1 and ms2 on Windows, and then run alphapept workflow to identified the extracted ms_data. I could get extractly the same identifications comparing with running the whole workflow on Windows. So the bug should be in pyrawfilereader. Same codes have different behaviors on Windows and MacOS.
Describe the bug
AlphaPept installation is cancelled after progress bar already reached the end. Security warning as shown in screenshot pops up. Error occurs both as user and administrator.
To Reproduce
Steps to reproduce the behavior:
Expected behavior
Installation should result in AlphaPept being installed.
Screenshots

Version (please complete the following information):
The current UI has several bugs, and several ideas for enhancement exists.
Currently, one can select FASTA files and a database file. This isn't very clear:
When are we creating a new database file, when are we using the existing one?
Several steps could become unresponsive when having extreme cases. Examples are when dropping thousands of files in the file dialog or when displaying first_search in the explore tab.
Possible Solutions:
Start the respective steps in a thread and show a wait indicator.
Probably because the fasta is too small to be divided into >1 fasta_block.
ValueError Traceback (most recent call last)
<ipython-input-3-cdeabb6ebdae> in <module>
1 from alphapept.runner import run_alphapept
----> 2 run_alphapept(params)
/home/feng/alphapept/alphapept/alphapept/runner.py in run_alphapept(settings, callback)
67 cb = callback
68
---> 69 spectra, pept_dict, fasta_dict = generate_database_parallel(settings, callback = cb)
70 logging.info('Digested {:,} proteins and generated {:,} spectra'.format(len(fasta_dict), len(spectra)))
71
/home/feng/alphapept/alphapept/alphapept/fasta.py in generate_database_parallel(settings, callback)
708 spectra_set.append(spectra[-1])
709
--> 710 pept_dict = merge_pept_dicts(pept_dicts)
711
712 return spectra_set, pept_dict, fasta_dict
/home/feng/alphapept/alphapept/alphapept/fasta.py in merge_pept_dicts(list_of_pept_dicts)
515
516 if len(list_of_pept_dicts) < 2:
--> 517 raise ValueError('Need to pass at least two elements to merge.')
518
519 new_pept_dict = list_of_pept_dicts[0]
ValueError: Need to pass at least two elements to merge.
Is your feature request related to a problem? Please describe.
In the "Settings" tab if a user looks at all available options and scroll them up and down and if at this moment the mouse cursor appears on one of the IntegetInput/FloatInput options, the values of these options could be changed even without the user's notice.
Describe the solution you'd like
To prevent the changing of the IntegetInput/FloatInput options in the "Settings" tab using the mouse scrolling.
Updating the github workflow before pushing a commit allows to run arbitrary code on the runners. While some changes require to modify the github workflow to actually make a CI pass, this can pose a serious security threat once the repository is publically accessible. How can/will we deal with this?
I revised the settings file to have some more consistency:
Please find attached the current set of options:
I roughly tried to sort the categories as follows:
calibration: Related to calibration
experiment: The experiment details
fasta: related to creating a theoretical database from fasta
features: related to feature finding
general: general workflow settings
misc: everything else
quantification: related to quantification options
raw: related to handling raw files
search: search options
All options are defined in the settings_template.yaml. This is what is used to create the user interface. It defines the type, some min/max and default values and also a brief description.
As there is always a lot of debate about the naming convention, I would be happy to collect ideas on which options should be renamed and which ones you find missing.
General idea
I would like to sugest a starting page for the GUI which briefly introduces AlphaPept and provides a mini-overview of what can be done and how. This can also be the place to provide a download button for the detailed user guide thats currently in the making. I think this would be good especially if people use AlphaPept for the first time and there is nothing to see or use on the 'status' page. What do you think?
Currently, dependable is not tracking the software versions.
Check why this is.
Ideally, going back to the requirements.txt would be nice.
The current route to create an installer on Windows is to use pyinstaller and inno setup.
While the pyinstaller script (create_installer.bat) runs through on some machines, it does not do work on the self-hosted runner.
In general, we should make an installation routine that is platform-specific as PyInstaller does not support cross-platform compilation.
Also, the current installer script create_installer.bat is not testing intermediate steps (e.g. was pyinstaller successful?) so we could have an exe installer that installs something that is not working, so intermediate testing steps are necessary.
This is particularly relevant for the GUI, as PyQT can cause problems, but a GUI is not that straightforward to test.
This issue will be linked to a project so that we can track the ToDo.
**- [ ] Loading of ext does not work -> Check installed version
Describe the bug
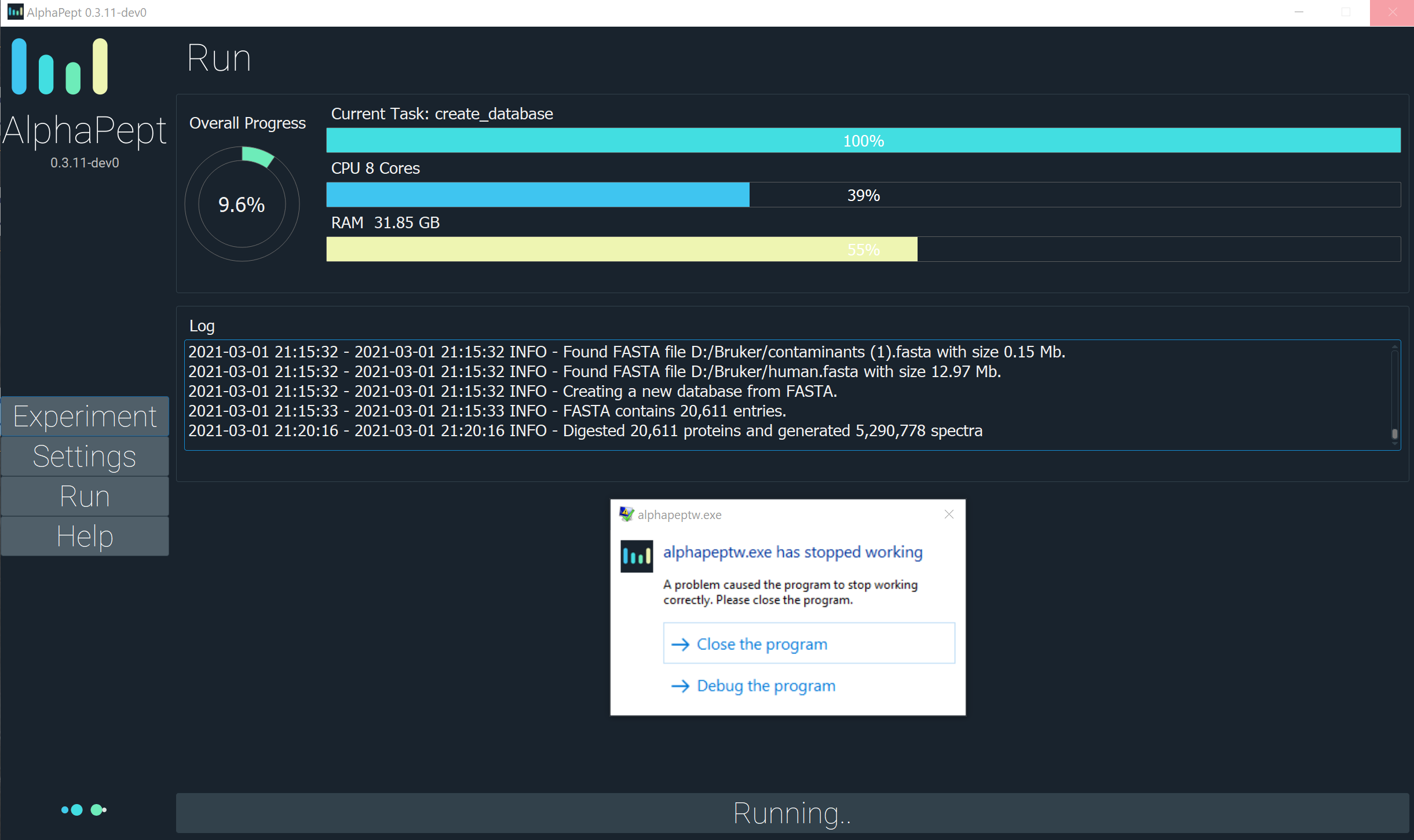
An unknown exception occurs when the process of the DB generation ends for the test HeLa file(Thermo) -
/04_hela_testrun/20190402_QX1_SeVW_MA_HeLa_500ng_LC11.raw.
To Reproduce
!!! After getting an exception, the process(es) of alphapept.exe still exists in the Task Manager like active.
settings.txt
Screenshots
https://i.gyazo.com/9b04df0db8a5c85c492e408928301c03.png
Version (please complete the following information):
Describe the bug
Not really a bug, but I would add units to the axes in the History tab of the GUI.
Specifically rt_length should be I guess in minutes and timing as well.
Hi,
The current git configs seem to be not in order. Please make sure that your git config is set correctly and you are using the correct credentials.

I am planning on re-writing the git history so that we all are consistent.
For now, I will change @ibludau and my username. @swillems and @jalew188 should I change yours as well? To the biochem email or the personal?
Dependabot can't evaluate your Python dependency files.
As a result, Dependabot couldn't check whether any of your dependencies are out-of-date.
The error Dependabot encountered was:
Illformed requirement ["==.0.71.0"]
To have a maintainable package, automated tests and performance benchmarks are crucial.
I have the following tests in mind, considering a versioning scheme where we are using (X-Y-Z): X-Major, Y-Minor, Z-Patch. In terms of branching we would have a master/dev and feature branches.
Simple function tests within nbdev. They should be run for every push on every branch. Duration ~ minutes
Tests to run a full pipeline (i.e., perform a search on HeLa Thermo / Bruker data). We could run them for every version, even minor on dev). Duration <1h
We would auto-create a settings template for the current version and replace the file_path with the respective filenames)
Test to try all possible (or at least most) combinations of settings.
This is something we could do for every Minor version. Duration will be several hours.
I think shipping is very crucial, and we should have one-click installers ready for each patch. To compile an installer takes approx. <10 minutes so this could be done for every push on the dev branch.
This is a very difficult test but very important to keep a userbase. Implementing a new feature and then the GUI doesn't work anymore. The current settings scheme is very flexible so that the core functionality should be tested with the Workflow test. A proper GUI test would probably include using tools like pyautogui that automatically "clicks" through workflows. If we want to be fancy, we could also use this to make a screenshoted documentation for each version automatically. Ideally I think this would be for every push on the dev branch.
The workflow test from above will not allow us to give a good estimate on performance. We will get execution time and proteins and peptides, but we should also consider metrics like quantification accuracy. For this, we should use multi-species samples with known mixing ratios that are computationally more demanding, and I would hence consider as a different kind of test.
The idea would be to have a set of PRIDE datasets like PXD010012, which we always re-run. As we could the analysis results from the repository, we would also have a baseline to compare the results to.
Depending on the number of datasets this could take
This is something we could potentially do for every minor version.
For running those tests, I will use GitHub-Actions self-hosted runners. This would allow us to use powerful workstations to run the tests.
Ideally, we can also set up runners for each Windows / Linux and Mac.
At some point, one could also make the testing results more explorable, i.e., pushing the results to a db and having a little dashboard app that shows performance over version/time.
Also, note that we can always trigger the tests manually.
Let me know if you would suggest additional or think the current test set should be optimized.
We should also add an automatic style test.
Hi,
After investigating the latest performance runs, I noticed that we have approx only 40k of peptides for Thermo Runs.
In the past, before the automated tracking, hen making the performance runs, we had approx. 50k, so it seems that we lost 20% somewhere...
I checked the settings, and they did not change (same FASTA / file, tolerances, etc.)
I presume that we have introduced a bug along the way (raw conversion? feature calibration? mapping MS1 to MS2) and should investigate.
Describe the bug
In GUI for all parameters in "Settings" tab that have a dropdown menu there is always an empty big space or one option from the menu is incomparable big.
To Reproduce
Can be reproduced for the following dropdown menus:
Expected behavior
It has to be the same size for all dropdown menu options.
Screenshots
https://i.gyazo.com/b2ef0ef36aa23bbb006e1aaf83d76404.jpg
https://i.gyazo.com/26d6244c747c468a7f1a86ee481a385a.jpg
Version (please complete the following information):
For the CI main, we apparantly use python3.6, while the sample files use python3.8.
This is very strange and turns out to mess up some things with the CI since dependencies might be different. Probably we should just make a single install script where e.g. a fixed conda setup (updates are relatively unreliable otherwise) installs python and alphapept consistenly. Calling this install script equally in all the .github/workflows should then be far more consistent.
Is your feature request related to a problem? Please describe.
The overall progress during DB generation starts only in several minutes after the running of the tool.
Describe the solution you'd like
Maybe, it would be better to show from the beginning 0.1% when the user just run the tool. Otherwise, it could look like that nothing happens. (see the screenshot - https://i.gyazo.com/cb1c9bb4d2721d4074147183881ce3a8.jpg)
There is a bug in the user interface, where the log is displayed. When having too many log updates in a short time, some characters appear to disappear. This seems to be related to the QTextEditLogger Widget.

This thread could be related: https://forum.qt.io/topic/76958/how-to-efficiently-display-almost-real-time-text-logging-without-freezing-an-ui
Is your feature request related to a problem? Please describe.
Some people use multiple proteases in one experiment. It would be cool if it were possible to also select multiple proteases in alphapept.

Describe the bug
I want to install alphapept on my Macbook with M1 chip (ARM). This does not work out-of-the-box. I want to share my learnings and open issues here for others to be more efficient in solving them with new dependency versions available.
Several packages will not work, such as numba, PyQt5 and pythonnet. One error message (regarding numba) is:
FileNotFoundError: [Errno 2] No such file or directory: 'llvm-config'
To Reproduce
Steps to reproduce the behavior:
conda create --name alphapept python=3.8
conda activate alphapept
cd
git clone https://github.com/MannLabs/alphapept.git
cd alphapept
pip install -r requirements.txt
pip install .Expected behavior
Installation runs through with out an error and within the python console, I can run:
import alphapeptVersion (please complete the following information):
Currently, the quantification accuracy with MaxLFQ is not yet satisfactory.
These are the results when running the species test from PXD010012

Notably, no clear distinction between the species is possible. The original data from the paper looks like this:

When testing only the algorithm part on the data, the following results can be observed.

The following two observations will be investigated:
To Reproduce
Expected behavior
To update the section when a file path is typed even if it's the same path.
Version (please complete the following information):
Is your feature request related to a problem? Please describe.
If you realise that you selected inappropriate settings or similar, there should be a possibility to cancel a job. Also, in case alphapept gets stuck (as happend for me - see below) you should be able to cancel.
Describe the solution you'd like
There could be a cancelation button in the 'queue' tab on the 'status' page.
Nice to have
There could also be an option to prioritise jobs in the queue. In case an urgent analysis comes up you could move it up in the list.
Additional context
Specifically, I have an issue with a job that (I guess) got stuck and it restarts and gets stuck at the same point again when I relaunch alphapept (this is stuck for >20 min already). I will write a separate issue for this.

There are quite some "stale" branches and some of the HDF branches were only merged and not properly deleted after merging/pulling.
I am not sure how it works if multiple people work on the ame branch, but ptobably we should try tro keep the github as clean as possible. The stale branches can probably be deleted, unless someone is acitvely working on it?
Merged/pulled branches can probably be deleted easily with a "merge and delete", kinstead of just merging, although I am not sure how good this is if other contributors are still working on this branch locally...
For what it is worth, I only consider the following branches active and all others can be deleted:
The NPZ format to store data files is only a temporary solution. @swillems has some fast implementations that use indexing and are being used in the ion networks project. This would allow to save even the query data in the hdf.
Keys steps would be:
In terms of architecture, several design ideas can be utilized.
So far, a major approach was to use numba-optimized functions for the core code. Numba allows to use OOP with Jitclasses. A downside here is that you need to type the variables, which affects the clear and easy python syntax.
As discussed, we could use a combination of having regular Python classes with Numba functions.
The UI implementation relies much more on OOP, and basically no numba functions are employed.
This issue is intended to collect ideas on where we should revise the code so that we have more flexibility for further modules.
Describe the bug
The timing plot is not correctly displayed in release 0.3.23-dev0
Screenshots

Version (please complete the following information):
Describe the bug
If terminate the analysis process at any point (f.e. FF) and after that try to rerun the analysis using default workflow steps,
"Processing of D:\04_hela_testrun\20190402_QX1_SeVW_MA_HeLa_500ng_LC11.ms_data.hdf for step raw_conversion failed. Exception File extension .hdf not understood." exception occurs in the terminal and the program is frozen.
To Reproduce
Steps to reproduce the behavior:

Expected behavior
It should be possible to rerun any terminated processes again.
As a suggestion, maybe, it makes sense not to show the .ms_data.hdf in the Raw files section.
Version (please complete the following information):
Attached log file.
2021_05_27_thermo_hela_run2.log
Is your feature request related to a problem? Please describe.
I think the setting selection is not optimal in a way that you cannot distinguish what is a setting selection and what opens a drop-down menu (see screenshot)

Here, 'raw' opens a drop-down selection, 'use_profile_ms1' is a setting and 'fasta' is a drop-down again.
Describe the solution you'd like
I would generally prefer if the overall drop-down categories wouldn't be selected by a tick box but rather with a plus on the right site as is done for the settings in general. Another option would to indent the actual settings selection or use another background color.
To Reproduce
Expected behavior
It would be nice to save the checking/unchecking of this check box during switching between the sections.
Version (please complete the following information):
Describe the bug
If you run a second analysis on the same folder as a previous analysis then the result files are overwritten. In the GUI you can however still select the yaml of the initial analysis, see the initial parameters, but non-matching results in the tables.
To Reproduce
Steps to reproduce the behaviour:
Expected behavior
I think it would be good to either append the name of the yaml file also to all result files as a general suffix for an analysis. This way alternative analysis results can be stored in teh same folder.
Alternatively, if only one set of results should be available per folder, I would suggest to restructure the results panel so that the results.yaml settings are shown and not any settings that were saved by the user at any point. They might not match the shown results.
Version (please complete the following information):
I used a wrong raw (DIA raw) file while testing, and a weird error showed below. It may be because some spectra are empty in the DIA run. We should check whether idxs_lower, idxs_higher are empty or not during the run?
ValueError Traceback (most recent call last)
in
1 from alphapept.runner import run_alphapept
----> 2 run_alphapept(params)
~/opt/anaconda3/lib/python3.8/site-packages/alphapept/runner.py in run_alphapept(settings, callback)
140 cb = callback
141
--> 142 fasta_dict, pept_dict = search_parallel_db(settings, callback=cb)
143
144 else:
~/opt/anaconda3/lib/python3.8/site-packages/alphapept/search.py in search_parallel_db(settings, calibration, callback)
1062 file_npz, settings_ = to_process[0]
1063 settings_['search']['parallel'] = True
-> 1064 search_db((file_npz, settings_))
1065 else:
1066 with Pool(n_processes) as p:
~/opt/anaconda3/lib/python3.8/site-packages/alphapept/search.py in search_db(to_process)
1022 features = pd.read_hdf(base+'.hdf', 'features')
1023
-> 1024 psms, num_specs_compared = get_psms(query_data, db_data, features, **settings["search"])
1025 if len(psms) > 0:
1026 psms, num_specs_scored = get_score_columns(psms, query_data, db_data, features, **settings["search"])
~/opt/anaconda3/lib/python3.8/site-packages/alphapept/search.py in get_psms(query_data, db_data, features, parallel, m_tol, m_offset, ppm, min_frag_hits, callback, m_offset_calibrated, **kwargs)
249 idxs_lower, idxs_higher = get_idxs(db_masses, query_masses, m_offset, ppm)
250 frag_hits = np.zeros(
--> 251 (len(query_masses), np.max(idxs_higher - idxs_lower)), dtype=int
252 )
253
<__array_function__ internals> in amax(*args, **kwargs)
~/opt/anaconda3/lib/python3.8/site-packages/numpy/core/fromnumeric.py in amax(a, axis, out, keepdims, initial, where)
2665 5
2666 """
-> 2667 return _wrapreduction(a, np.maximum, 'max', axis, None, out,
2668 keepdims=keepdims, initial=initial, where=where)
2669
~/opt/anaconda3/lib/python3.8/site-packages/numpy/core/fromnumeric.py in _wrapreduction(obj, ufunc, method, axis, dtype, out, **kwargs)
88 return reduction(axis=axis, out=out, **passkwargs)
89
---> 90 return ufunc.reduce(obj, axis, dtype, out, **passkwargs)
91
92
ValueError: zero-size array to reduction operation maximum which has no identity
For phosphorylation as an example, there may be many phospho sites on a sequence, resulting in a lot of isoforms. If max_isoforms is large enough, there may be too many isoforms to be considered; if max_isoforms is too small, modifications on left-side AAs may be excluded due to the behavior of itertools.product.
seq = "SBCDMFSSSSSSSSSSSMMFD" # maybe more phosites when sequence gets longer
mod_dict = dict(zip("S,M".split(","), "(ph)S,(ox)M".split(",")))
max_isoforms = 1000000
peplist = []
from time import perf_counter
start = perf_counter()
for i in range(1000):
peptides = get_isoforms(mod_dict, seq, max_isoforms)
peplist.extend(peptides)
end = perf_counter()
print(peptides[:100])
import sys
print(f'Memory usage and running time for 1000 repeats: {sys.getsizeof(peplist)/10**6:2f} MB, {end-start:2f} s, # of isoforms without repeat: {len(peptides)}')
Outputs:
['SBCDMFSSSSSSSSSSSMMFD', 'SBCDMFSSSSSSSSSSSM(ox)MFD', 'SBCDMFSSSSSSSSSSS(ox)MMFD', 'SBCDMFSSSSSSSSSSS(ox)M(ox)MFD', 'SBCDMFSSSSSSSSSS(ph)SMMFD', 'SBCDMFSSSSSSSSSS(ph)SM(ox)MFD', 'SBCDMFSSSSSSSSSS(ph)S(ox)MMFD', 'SBCDMFSSSSSSSSSS(ph)S(ox)M(ox)MFD', 'SBCDMFSSSSSSSSS(ph)SSMMFD', 'SBCDMFSSSSSSSSS(ph)SSM(ox)MFD', ..., 'SBCDMFSSSSSS(ph)SS(ph)S(ph)S(ph)S(ox)MMFD', 'SBCDMFSSSSSS(ph)SS(ph)S(ph)S(ph)S(ox)M(ox)MFD', 'SBCDMFSSSSSS(ph)S(ph)SSSSMMFD', 'SBCDMFSSSSSS(ph)S(ph)SSSSM(ox)MFD', 'SBCDMFSSSSSS(ph)S(ph)SSSS(ox)MMFD', 'SBCDMFSSSSSS(ph)S(ph)SSSS(ox)M(ox)MFD']
Memory usage and running time for 1000 repeats: 271.614064 MB, 14.134513 s, # of isoforms without repeat: 32768
Describe the bug
If I deinstall AlphaPept this does not delete the .alphapept folder.
Should this be the case?
A declarative, efficient, and flexible JavaScript library for building user interfaces.
🖖 Vue.js is a progressive, incrementally-adoptable JavaScript framework for building UI on the web.
TypeScript is a superset of JavaScript that compiles to clean JavaScript output.
An Open Source Machine Learning Framework for Everyone
The Web framework for perfectionists with deadlines.
A PHP framework for web artisans
Bring data to life with SVG, Canvas and HTML. 📊📈🎉
JavaScript (JS) is a lightweight interpreted programming language with first-class functions.
Some thing interesting about web. New door for the world.
A server is a program made to process requests and deliver data to clients.
Machine learning is a way of modeling and interpreting data that allows a piece of software to respond intelligently.
Some thing interesting about visualization, use data art
Some thing interesting about game, make everyone happy.
We are working to build community through open source technology. NB: members must have two-factor auth.
Open source projects and samples from Microsoft.
Google ❤️ Open Source for everyone.
Alibaba Open Source for everyone
Data-Driven Documents codes.
China tencent open source team.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}