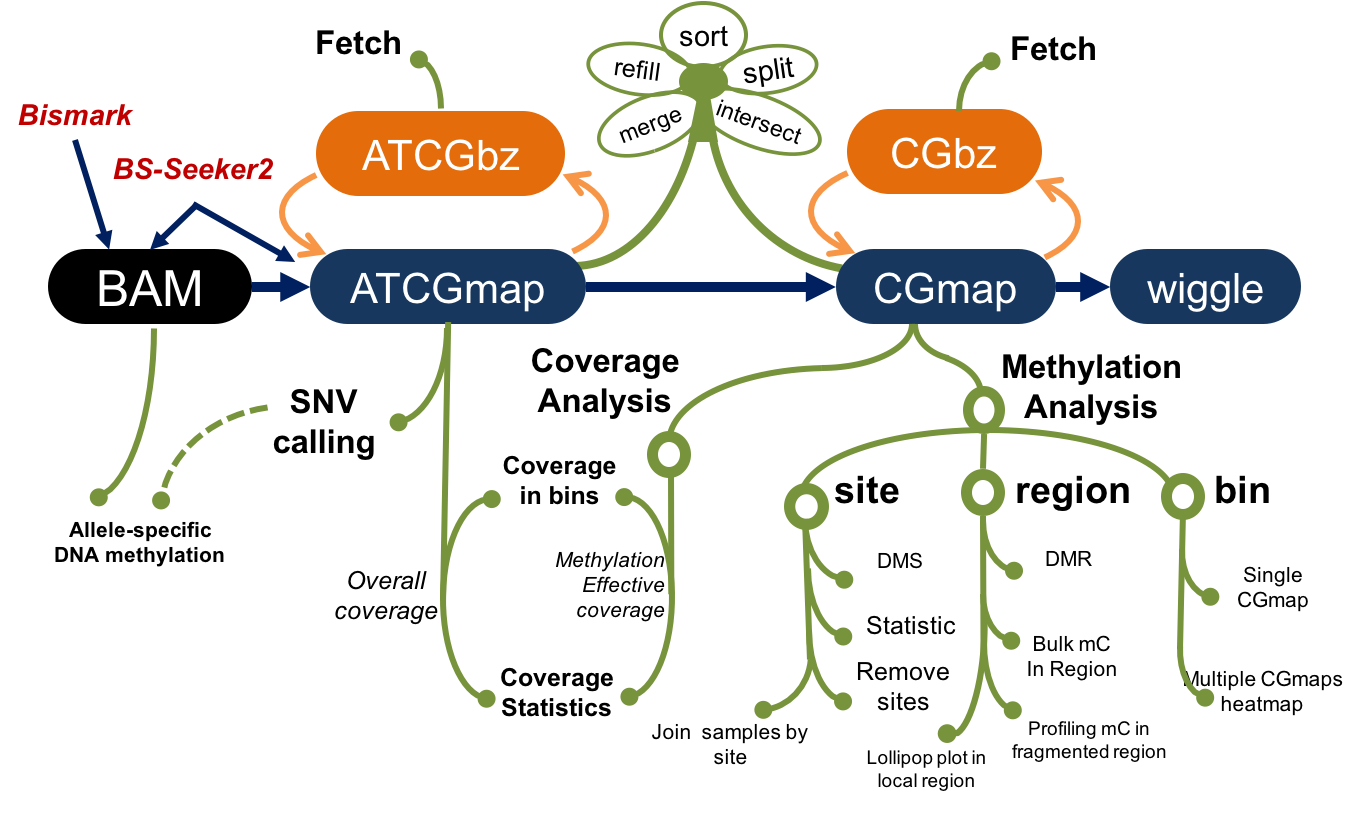
Command-line Toolset for Bisulfite Sequencing Data Analysis
homepage: https://cgmaptools.github.io
toolbox for analysing BS-seq data, advance features in SNV, ASM and DMR
Home Page: https://cgmaptools.github.io
Command-line Toolset for Bisulfite Sequencing Data Analysis
homepage: https://cgmaptools.github.io































































Dear Weilong,
I'm very happiness to find your nice software and paper.
In my own work, I have one question about the methylation level value.
I use this command to get Allele-specific DNA methylation site:
cgmaptools asm -m ass -r Chr12.fa -b Chr12.bam -l Chr12q.vcf -o Chr12.asm -t C
This is my result text:
Chr SNP_Pos Ref Allele1 Allele2 C_Pos Allele1_linked_C Allele2_linked_C Allele1_linked_C_met Allele2_linked_C_met pvalue fdr ASM
Chr12 9820 G G A 9814 3-3 5-0 0.50 1.00 1.82e-01 4.03e-01 FALSE
Chr12 9820 G G A 9869 7-0 5-0 1.00 1.00 1.00e+00 1.00e+00 FALSE
Chr12 9820 G G A 9897 7-0 5-0 1.00 1.00 1.00e+00 1.00e+00 FALSE
Chr12 9820 G G A 9901 7-0 5-0 1.00 1.00 1.00e+00 1.00e+00 FALSE
Chr12 9820 G G A 9908 7-0 4-0 1.00 1.00 1.00e+00 1.00e+00 FALSE
Chr12 9820 G G A 9922 7-0 4-0 1.00 1.00 1.00e+00 1.00e+00 FALSE
Chr12 9826 G G A 9814 3-3 5-0 0.50 1.00 1.82e-01 4.03e-01 FALSE
Chr12 9826 G G A 9869 8-0 5-0 1.00 1.00 1.00e+00 1.00e+00 FALSE
Chr12 9826 G G A 9897 8-0 5-0 1.00 1.00 1.00e+00 1.00e+00 FALSE
Chr12 9826 G G A 9901 8-0 5-0 1.00 1.00 1.00e+00 1.00e+00 FALSE
Chr12 9826 G G A 9908 8-0 4-0 1.00 1.00 1.00e+00 1.00e+00 FALSE
Chr12 9826 G G A 9922 8-0 4-0 1.00 1.00 1.00e+00 1.00e+00 FALSE
Chr12 9830 G G A 9814 3-3 5-0 0.50 1.00 1.82e-01 4.03e-01 FALSE
Chr12 9830 G G A 9869 9-0 7-0 1.00 1.00 1.00e+00 1.00e+00 FALSE
Chr12 9830 G G A 9897 9-0 7-0 1.00 1.00 1.00e+00 1.00e+00 FALSE
Chr12 9830 G G A 9901 9-0 7-0 1.00 1.00 1.00e+00 1.00e+00 FALSE
Chr12 9830 G G A 9908 9-0 6-0 1.00 1.00 1.00e+00 1.00e+00 FALSE
Chr12 9830 G G A 9922 9-0 6-0 1.00 1.00 1.00e+00 1.00e+00 FALSE
But I find some sites are repetitive and have different allele methylation level beacuse of basing on the different SNV. For now, if i want to know the site's (e.g 9901) two allele absolute methylation level. How can I get it ?
Best,
Jiaming
Hi Guo,
sorry to disturb you again, but I still have some questions. I used cgmapstat to obtain relative information with the rice.CGmap.gz as input. The result is as the picture show:

In the plant genome, we always pay attention to CG, CHH, CHG, and in the column 4 of CGmap.gz, it also only have three classes of DNA methylation, so how the CA, CT, CC, CW..... in the *.mstat.log come? They are not belong to CHH or CHG?
I used zcat CGmap.gz| awk '$4=="CG"' >CG.txt to count the CG (CHG, CHH)methylation sites, but if in the CG.map.gz there are also CA, CT... how could I get the accurate CG ,CHG and CHH sites?
Thank you for your patient reply. Zhang
Hi,
I run the tool to convert the BSSeeker2 produced bam to cgmap. It used all the memory of my machine (64GB) and cannot proceed generating truncated output.
"gzip: WGBS.CGmap.gz: unexpected end of file"
bam file size is 14G, ~380 million records. Does the tool demands lot of memory?
Thanks,
Hanbin
Hi,
Thanks for the great set of tools but I am having an issue with CGmapMethInBins.
My CGmap file looks like this:
chr1 C 10004 CHH CC 0.0 0 1
chr1 C 10005 CHH CC 0.0 0 1
However when I run the command :
cgmaptools mbin -i head_chr1.CGmap.txt -c 1 -B 25 > 25bin.tsv
The output has na where there should be 0 as in the CGmap above:
chr1 10001 10025 na
Also the resultant file does not even cover all of the positions in the input file.
tail -n 5 head_chr1.CGmap
chr1 C 264050 CHG CA 0.0 0 1
chr1 C 264057 CHH CA 0.0 0 1
chr1 C 264059 CHH CA 0.0 0 1
chr1 C 264061 CHH CA 0.0 0 1
chr1 C 264078 CHH CA 0.0 0 1
tail -n 5 25bin.tsv
chr1 121301 121325 na
chr1 121326 121350 na
chr1 121351 121375 na
chr1 121376 121400 na
chr1 121401 121425 na
This cropping is even worse when using context specific mbin.
cgmaptools mbin -i head_chr1.CGmap.txt -c 1 -B 25 -C CG > 25CGbin.tsv
tail -n 5 25CGbin.tsv
chr1 13676 13700 na
chr1 13701 13725 na
chr1 13726 13750 na
chr1 13751 13775 na
chr1 13776 13800 na
cgmaptools Version: 0.1.2
I have tried with python 2.6.9 and python 3.7.2 and the problem appears in both.
I have attached the sample of my CGmap file that I used in the above examples
Any help with this would be greatly appreciated.
Hello there,
I'm running bam2cgmap to get a cgmap file from a bam file.
However, when I ran: cgmaptools convert bam2cgmap -b 1.bam -g ge.fa --rmOverlap -o 1_cgmap,
I got: /software/cgmaptools-master/bin/CGmapFromBAM: /lib64/libz.so.1: version `ZLIB_1.2.3.5' not found (required by /software/cgmaptools-master/bin/CGmapFromBAM).
I wonder how to figure this out? Do I need to install zlib1.2.3.5 by myself (I have zilb1.2.11 already)? If so, where the path should it be installed?
Thanks!
Hi Dr. Guo,
When I tried to do the MTR analysis, I find out result as followed:
1 21800 21991 NA 0 NA 0
But when I trace back to the CGmap.gz file, I can clearly see that there is methyaltion there:
1 C 21823 CG CG 1.0 1 1
1 G 21824 CG CG 1.0 2 2
1 C 21845 CG CG 1.0 1 1
1 G 21846 CG CG 1.0 2 2
1 C 21849 CG CG 1.0 2 2
1 G 21850 CG CG 0.5 1 2
1 C 21851 CG CG 1.0 2 2
1 G 21852 CG CG 1.0 2 2
1 C 21853 CG CG 1.0 2 2
1 G 21854 CG CG 0.0 0 2
1 C 21882 CG CG 1.0 1 1
1 G 21883 CG CG 1.0 1 1
1 C 21897 CG CG 1.0 2 2
1 C 21912 CG CG 1.0 2 2
1 C 21914 CG CG 1.0 2 2
1 C 21926 CG CG 1.0 2 2
1 C 21939 CG CG 1.0 2 2
1 C 21953 CG CG 1.0 2 2
1 C 21975 CG CG 1.0 4 4
The command I used for the analysis is:
cgmaptools mtr -i rep3CHG.gz -r ${MITE} -o B73_rep3_CHG_MITE.mtr
Would you please tell me how can I fix this problem?
Yibing
Hi
How can I use cgmaptools for paired-end reads that aligned by bismark? At the moment cgmaptools don't support it.
Regards
Would it be possible to make environment dependencies (python2) and data input requirements (sorted bam) explicit in the documentation?
Also I would have expected the tool to produce a halting error, but currently the error is just logged and the whole workflow is allowed to continue (i.e. empty files are generated).
When running ASM, you can set the C depth as a command line option.
In the code for ASM.pl, when the number is set to 0, there is an error message on line 126 that says "Error: Minimum read depth for C site to call methylation level when calling asr should be no less than 1".
However, if you actually set the option to 0 on the command line, then on line 110, there is an assignment operator that will automatically re-set to the default value of 3 (since the value of "0" is read as "FALSE", it will over-write the value).
The script does not tell you that it does this, and the error message is never printed. So the output is the same as if the default was set to 3, but it does not warn you about this!
I believe the same issue will happen when setting any of the parameters to 0, since the operator "||=" will always read the left hand side as "FALSE".
The operators should be changed to only set default values when the parameters have not been specified on the command line.
Hello Weilong,
I have been using cgmaptools to analyze my wgbs reads following bsseeker2 pipeline. I ran into a hiccup while determining coverage in the CGmap file.
I used the following code:
cgmaptools mec stat -i ~/scratch/Align/MethCalls/CGmap/1117IL.fm.sort.rmdup.bam.CGmap.gz.CGmap -p 1117IL -f png > 1117IL.mec_stat.data
The code ran to completion and I get the 1117IL.mec_stat.data file; however, it did not produce figures. I received following error message
ValueError: Image size of 1100x140976 pixels is too large. It must be less than 2^16 in each direction.
It seems like the default value of pixel is too large; is there an easy fix to this problem? I would prefer to have those figures depicting methylation coverage.
Thank you,
Suraj
Hello,
As the data of WGBS is huge.
I have tested bismark for alignment. But it took lots of time.
Currently, like biscuit, bwameth can finish the alignment in more short time.
So, could cgmaptools support bam file produced by biscuit or bwameth?
Thanks for your attention!
I noticed something interesting when converting bam files generated from BSseeker2 into CGmap's using Bam2CGmap function. When I do NOT specify --rmOverlap, coverage falls around 9000 , which is what I expected. When I DO specify --rmOverlap, coverage seems to be pushed down to a maximum 1000. Why and is there a way to rectify this? I would like to retain as much coverage as possible.
Unrelated, when I use BSseeker2 bam to cgmap tool, coverage falls to the tens or hundreds. This is incredibly frustrating, which is why I prefer CGmaptools over BSseeker2 for methylation calling.
Thank you,
Kristin
I run Bismark, and then cgmaptool to look for DMR between two conditions
I got a DMR file after intersecting two files obtained after using cgmaptools, but I do not fully understand or know how to interpret such a DMR file
e.g. This first column is part of the final DMR file (used in a CG context)
Fvb1 1638 1695 58 -1.4065 1.90e-01 0,9483 1 0,9483 6
I understand the under condition 1, 94,83% of the 6 CG are methylated, whereas 100% are methylated under condition 2.
If that is true, how and why this is recognized as a DMR?. Should I apply a p-value filtering ?

Hi. Typically the Watson strand C and Crick strand C will be listed separately in the CGmap file. But I prefer to combine them into a single position (by summing the read counts) for visualization in the genome browser. Does cgmaptools have a way to do this? The most intuitive place would be in the cgmap2wig function, but I don't think it supports that. Is there a way to do it?
Thanks! Ben.
Hi, Weilong.
I find the size of my SNV file is 19G from 40G bam file with your software cgmaptools. And I want to predict ASM regions from my SNV file with cgmaptools asm. But I find that its process is very slow and it can't generate asm file or its size is zero. And I tried to test with SNV in 1G size, it did work.
Question1: can you change your code about cgmaptools asm in order to use multiple threads and cpus' parameter?
Question2: my SNV file could be splited into several parts based on chromosome name, and I do the cgmaptools asm part. Then merge these asm file. Is it right? Is its FDR right?
Thanks very much.
Looking forward to your reply.
Best wishes.
Grace
The following conversion gives trancated gz file.
cgmaptools convert bam2cgmap -b ${SAMPLE}_merged_final_sorted_dedup.deduplicated.bam -g ${GENODIR}/hg38_r87.fa --rmOverlap -o ${SAMPLE}_cgmap
The sorted bam file has 10GB in all cases the script completed without any problem, shows # processing reads from: chr1 - MT, BUT the final ${SAMPLE_cgmap.CGmap.gz file stop on chr X with a truncated end. NO chr Y and MT. This is a bismark alignment.
Best,
@demis001
Hi WeiLong:
I have a question about the program.
If I remove the code " # next if ( $allele1 eq $allele2 ); # only retain heterozygous SNP site (htSNP)" of ASM.pl, the results will contain the methylation information of homozygous SNP sites. Are the results correct?
Thanks
Best
Hello,
I'm trying to use cgmaptools to analyze my methylation data. My data is huge (~60Gb bam file per sample, 9 samples with 2 sets of treatment and 1 set of control). I failed to call the methylation using CGmapFromBAM -O option (to remove overlap). This issue has been reported and hope it can be solved soon.
I finally used MethylDackel to call the methylation from bam files and converted them to CGmaps. But each CGmap is still ~40Gb in size. I tried to run methylKit and DSS for the analysis but R is terrible at processing big data, so I turn to CGmaptools. I haven't finished the analysis yet, but seems it still takes a while to processing these samples using CGmapStatCov (not as RAM-consuming as R). Can I run my samples on cgmaptools parallelly by allocating more CPUs?
BTW, seems that I can only merge my replicates to do the differential methylation analysis on cgmaptools. Why not using the replicates information to do the statistics inference. Is there an evaluation/comparison on different statistical models used for the DM analysis?
Thanks
Ziliang
Hi,
I'm trying to use cgmaptools mtr to call methylation in multiple regions, but I get the following error:
cgmaptools mtr -i sample.CGmap.gz -r regions.bed
Traceback (most recent call last):
File "/Users/gcristof/Lab/bioinfo/tools/cgmaptools/bin/CGmapToRegion", line 211, in <module>
main()
File "/Users/gcristof/Lab/bioinfo/tools/cgmaptools/bin/CGmapToRegion", line 205, in main
CGmapToRegion(options.CGmapFile, options.regionFile)
File "/Users/gcristof/Lab/bioinfo/tools/cgmaptools/bin/CGmapToRegion", line 102, in CGmapToRegion
key_c = Get_key(chr_c)
File "/Users/gcristof/Lab/bioinfo/tools/cgmaptools/bin/CGmapToRegion", line 38, in Get_key
match = re.match(r"^chr(\d+)", str, re.I)
File "/opt/local/Library/Frameworks/Python.framework/Versions/3.7/lib/python3.7/re.py", line 173, in match
return _compile(pattern, flags).match(string)
TypeError: cannot use a string pattern on a bytes-like object
Curiously, when I first zcat the sample.CGmap.gz file and pipe the result to cgmaptool mtr, it starts to output some lines, then from chr2, I get plenty of NA lines, and finally I get another error:
zcat sample.CGmap.gz | cgmaptools mtr -r regions.bed
Traceback (most recent call last):
File "/Users/gcristof/Lab/bioinfo/tools/cgmaptools/bin/CGmapToRegion", line 211, in <module>
main()
File "/Users/gcristof/Lab/bioinfo/tools/cgmaptools/bin/CGmapToRegion", line 205, in main
CGmapToRegion(options.CGmapFile, options.regionFile)
File "/Users/gcristof/Lab/bioinfo/tools/cgmaptools/bin/CGmapToRegion", line 104, in CGmapToRegion
if key_c < key_r :
TypeError: '<' not supported between instances of 'int' and 'str'
I suspect a problem of sorting, but I get similar error with the cgmaptools sort command. In the help message it is indicated:
Note: The two input CGmap files should be sorted by Sort_chr_pos.py first.
What are the 2 input files? I only have one. And where can I find the Sort_chr_pos.py script ? not in cgmap toolbox?
Thanks for your help!
i use cgmaptools to call snv,and show the file may have wrong number of columns
command
cgmaptools snv -i SRR306438.CGmap.gz -m bayes -v SRR306438.bayes.vcf -o SRR306438.snv --bayes-dynamicP
cgmap.gz
chr1 C 3146 CG CG 1.0 1 1
chr1 C 3157 CHH CC 0.0 0 1
chr1 C 6281 CHH CT 0.0 0 1
chr1 G 36957 CHG CT 0.0 0 1
chr1 G 46814 CHH CA 0.0 0 1
chr1 G 57453 CHH CA 0.0 0 1
chr1 G 66407 CHH CC 0.0 0 1
chr1 G 73910 CHH CT 0.0 0 1
chr1 G 77600 CHG CT 0.0 0 1
chr1 G 77628 CHH CC 0.0 0 1
chr1 G 78179 CG CG 0.0 0 1
chr1 G 81834 CHG CA 0.0 0 1
chr1 C 91607 CHH CA 0.0 0 2
chr1 C 91633 CHH CA 0.0 0 2
chr1 C 96617 CHH CT 0.0 0 1
chr1 C 123259 CHH CC 0.0 0 1
chr1 C 127602 CHG CA 0.0 0 1
chr1 C 222294 CHG CT 0.0 0 1
@guoweilong Hi, I tried to obtain atcgmap.gz cgmap.gz from Bismake bam file with 'cgmaptools convert bam2cgmap' command, but always reported the error 'free(): invalid pointer' as shown below. Finally, no atcgmap.gz file was generated or the generated ATcgmap.gz file was not complete enough for subsequent analysis.
free(): invalid pointer
Would you please tell me how can I fix this problem? Thank you very much
Han Xu
Hi:
I've got some VCF files from the SNV calling. However, no genotype is 0/0. Is it a correct result?
Thanks
DMR out file:
1 13216 13303 -4.0234 2.43e-03 0.7300 0.9733 6
Intersect file:
1 C 13216 CG CG 0.62 5 8 0.90 9 10
1 G 13217 CG CG 1.00 5 5 1.00 4 4
1 C 13283 CG CG 0.76 13 17 1.00 21 21
1 G 13284 CG CG 0.70 7 10 1.00 10 10
1 C 13302 CG CG 0.63 12 19 1.00 22 22
1 G 13303 CG CG 0.67 10 15 0.94 17 18
This looks 3 CG site to me? Any idea, how the number DMR number generated?
Hi WeiLong:
I have a question about the ASM.
What format of VCF info should I provide for ASM.pl, is the Bis-SNP output file works? if only heterozygous SNP site used for ASM calling? And how is it works when I don`t have a SNP file?
And what was advanced of SNV calling, which method should I select.
I have run it for >24h with no output..
Best Han
2017/11/29
#command:
cgmaptools mergelist tomatrix -i index -f TS559exoS.CGmap,TS559statS.CGmap -t exoS,statS -o TS559_mC_map -c 20
#head of index file:
TS559_Genomic_Sequence 6
TS559_Genomic_Sequence 7
TS559_Genomic_Sequence 9
TS559_Genomic_Sequence 12
TS559_Genomic_Sequence 14
TS559_Genomic_Sequence 18
TS559_Genomic_Sequence 21
TS559_Genomic_Sequence 26
TS559_Genomic_Sequence 27
TS559_Genomic_Sequence 40
error message:
File "/installs/cgmaptools-master/bin/CGmapFillIndex", line 119
print "\t".join( [pos] + Site[pos] )
^
SyntaxError: invalid syntax
#i've also tried gzipping the index and CGmaps files
Dear Guo,
recently I tried cgmaptools for methylation analysis, I used bismark for rice methylation extractor (pair-end), and then used cgmaptools convert bismark2cgmap to generate a CGmap file, then I want to generate a genomewide methylation level by cgmaptools mbin with the command: zcat 2n-1M.CGmap.gz | cgmaptools mbin -B 10000 -c 5 -f pdf -p 2n-1M.mbin -t 2n-1M > 2n-1M.mbin.log, finally get the pdf as below:

but my pdf doesn't seem to be a right profile as I saw in a user guide, is there something wrong with my process? And I see the
order of the chromosome is not like "Chr1, Chr2, Chr3...", how can I adjust the order ?I want to use cgmaptools mbin to divide the genome to some bins then used the mean methylation level of each bin to caculate the correction between replicates, is it feasible? and what is the appropriate length of the bins?
Waiting for your reply, and thanks a lot.
Hi,
Thanks for what looks like a great package. In the documentation for for the lollipop plot, you suggest the example
cgmaptools lollipop -i matrix.CG.gz -a anno.refFlat
But theres no information on how to use the -a flag or the format for anno.refFlat. Can you provide some more documentation on how you obtained the example lollipop plot?
Thanks,
Dear Weilong,
Hello. I used BS-seeker2 to align RRBS data and created CGmap files. But I got an error below when starting to run cgmaptools intersect. Without gzipped file, there was no error.
gblab@gblab:/backup/projects/RRBS/bsSeeker/methyl_call$ cgmaptools intersect -1 liver.CGmap.gz -2 muscle.CGmap.gz -o test.gz
Traceback (most recent call last):
File "/home/gblab/programs/cgmaptools/bin/CGmapIntersect", line 184, in <module>
main()
File "/home/gblab/programs/cgmaptools/bin/CGmapIntersect", line 179, in main
CGmapIntersect(options.CGmap_1, options.CGmap_2, options.context)
File "/home/gblab/programs/cgmaptools/bin/CGmapIntersect", line 129, in CGmapIntersect
b"%.2f" % float(methyl_2), b"%d" % int(NmC_2), b"%d" % int(NC_2)]) )
TypeError: sequence item 0: expected str instance, bytes found
Can you please help me solve this error? Though I can gunzip every CGmap.gz and use CGmaptools Intersect for now, I want to use gz file next time.
Best,
ByeongYong.
Dear Weilong,
I want to draw the heatmap like Figure 7.3. Firstly, I ran the command as follows,
cgmaptools mmbin -l 1.CGmap.gz,2.CGmap.gz,3.CGmap.gz > mmbin.tab
#cgmaptools heatmap -i mmbin.tab -c -o cluster.pdf -f pdf
and get ERROR,
File "/mnt/duankui/soft/cgmaptools-0.1.2/bin/CGmapsMethInBins", line 238, in
main()
File "/mnt/duankui/soft/cgmaptools-0.1.2/bin/CGmapsMethInBins", line 232, in main
int(options.bin_size), options.context )
File "/mnt/duankui/soft/cgmaptools-0.1.2/bin/CGmapsMethInBins", line 165, in CGmapMethylInBins
line = IN.readline()
File "/usr/local/anaconda3/envs/pyscenic/lib/python3.7/gzip.py", line 385, in readline
return self._buffer.readline(size)
File "/usr/local/anaconda3/envs/pyscenic/lib/python3.7/_compression.py", line 68, in readinto
data = self.read(len(byte_view))
File "/usr/local/anaconda3/envs/pyscenic/lib/python3.7/gzip.py", line 493, in read
raise EOFError("Compressed file ended before the "
EOFError: Compressed file ended before the end-of-stream marker was reached
so, any suggestion can be given to fix the problem!
Thank you for your generous help!
@guoweilong
Dear Dr. Guo,
Hello! I ran bam2cgmap to get a cgmap file from a deduplicated bam file generated by Bismark. The command line was: 'cgmaptools convert bam2cgmap -b xxx.deduplicated.bam -g xxx.fas --rmOverlap -o ./XXX/'
But the log file reported that:
input bam file: xxx.deduplicated.bam
source genome file: xxx.fas
[selected mode] Remove overlap
prefix for output CGmap: ./XXX/
[bam_pileup_core] the input is not sorted (reads out of order)
[bam_plp_destroy] memory leak: 1. Continue anyway.
After I used samtools to sort the bam file by read name or not use the parameter '--rmOverlap', the same error information was still reported. May i have your suggestion for this issue?
Thanks!
@guoweilong @benbfly @Perry-Zhu @Miaolingfeng
I converted CGmap to wig using the following command and got negative ratio in the wig format. Would you please tell me what does that mean?
cgmaptools convert cgmap2wig -i oocyte_merged_sorted_dedup_CGcontext_updated.CGmap -w oocyte_merged_sorted_dedup_CGcontext_updated_new.wig -c 1 -b 0
CGMP file
`
head -n 1000 oocyte_merged_sorted_dedup_CGcontext_updated.CGmap
1 C 10469 CG CG 0.00 0 3
1 C 10525 CG CG 0.00 0 1
1 C 10542 CG CG 0.00 0 1
1 C 10563 CG CG 0.00 0 1
1 C 10571 CG CG 0.00 0 1
1 C 10577 CG CG 0.00 0 1
1 C 10579 CG CG 0.00 0 1
1 C 10589 CG CG 0.00 0 1
1 C 10696 CG CG 0.00 0 1
1 C 10699 CG CG 0.00 0 1
1 C 10702 CG CG 0.0 0 2
1 C 10708 CG CG 0.0 0 2
1 C 10718 CG CG 0.0 0 2
1 C 10720 CG CG 0.0 0 2
1 C 10723 CG CG 0.0 0 2
1 C 10725 CG CG 0.0 0 2
1 C 10728 CG CG 0.0 0 2
1 C 10731 CG CG 0.0 0 2
1 C 10737 CG CG 0.0 0 2
1 C 10747 CG CG 0.0 0 2
1 C 10749 CG CG 0.0 0 2
1 C 10752 CG CG 0.0 0 2
1 C 10754 CG CG 0.0 0 2
1 C 10757 CG CG 0.0 0 2
1 C 10760 CG CG 0.0 0 2
1 C 10766 CG CG 0.0 0 2
1 C 10776 CG CG 0.0 0 2
1 C 10778 CG CG 0.0 0 2
1 C 10781 CG CG 0.0 0 2
1 C 10783 CG CG 0.0 0 2
1 C 10786 CG CG 0.0 0 2
1 G 10790 CG CG 0.00 0 1
1 G 13284 CG CG 1.00 1 1
1 G 13303 CG CG 1.00 1 1
1 C 14349 CG CG 1.00 1 1
1 G 15644 CG CG 1.00 1 1
1 G 16244 CG CG 1.00 1 1
1 C 16822 CG CG 1.00 2 2
1 G 16963 CG CG 1.00 1 1
`
Wig file:
`
head -n 1000 oocyte_merged_sorted_dedup_CGcontext_updated_new.wig
variableStep chrom=1
10469 0.00
10525 0.00
10542 0.00
10563 0.00
10571 0.00
10577 0.00
10579 0.00
10589 0.00
10696 0.00
10699 0.00
10702 0.00
10708 0.00
10718 0.00
10720 0.00
10723 0.00
10725 0.00
10728 0.00
10731 0.00
10737 0.00
10747 0.00
10749 0.00
10752 0.00
10754 0.00
10757 0.00
10760 0.00
10766 0.00
10776 0.00
10778 0.00
10781 0.00
10783 0.00
10786 0.00
10790 -0.00
13284 -1.00
13303 -1.00
14349 1.00
15644 -1.00
16244 -1.00
16822 1.00
16963 -1.00
`
Best,
@demis001
First, It's a good software for DNA methylation analysis. but When I run the command 'cgmaptools mbin -i WG.CGmap.gz -B 500 -c 4 -f png -t WG -p WG > mbin.WG.data' , but display 'File "/public/home/qliu/bio_soft/cgmaptools/bin/CGmapMethInBins", line 66
print "\n[Error]:\n\t File cannot be open: ", fn' . I open the script of CGmapMethInBins, find line 66 was
'print "\n[Error]:\n\t File cannot be open: ", fn'。 but it use in python2 not in python3. finally I fix the conmmand with ''print("\n[Error]:\n\t File cannot be open: ", fn) " ,and then run without mistake report.
Hi there--I've tried to run mstat numerous times now on .CGmap and .CGmap.gz, but it fails despite multiple iterations. Running the latter (.gz format) generates an "IOError: CRC check failed" in gzip.py, while running the .CGmap version generates this error: "File [ .CGmap ] may have wrong number of columns." I've tried running with and without figure-generation, but it keeps hitting these errors. Any thoughts on what I could do to fix this? Thanks so much for building this wonderful tool!!
Hello, guoweilong! Thank you for your tool!
I tried to call SNVs from ATCGmap file with 'bayes' option as in the tutorial:
cgmaptools snv -m bayes --bayes-dynamicP -i file1.ATCGmap -v file1.vcf
and got the error:
File "SNVFromATCGmap.py", line 493, in PredictNT_bayes
[Prob, geno] = max(zip(pstP, ClearSet) + zip(V_pstP, VagueSet) )
TypeError: unsupported operand type(s) for +: 'zip' and 'zip'
Used python v 3.8.5. I suggest changing zips to lists in order to join them.
Hi, WeiLong:
I have sequenced some single-cell bisulfite PE data, and aligned to reference using bismark.
In your related scBS-seq paper, you also used the unmapped single reads for mapping.
In my study, I had used the --non_directional for both PE mapping and unmapped single reads mapping, and BAM files was merged by using samtools merge, and followed by duplication by samtools in SE mode( -s and -S, for both PE and SE in the merged SAM file ). And bismark_methylation_extractor was used for mC calling.
So, was that rigth ? My quenstion is that Mr FuChou alway use that umapped single end reads for alignement, however if the PE and SE alignment results could result ambiguous mC.
Look forward your reply! And glad for your suggestion.
Thank you very much !
New Paper comming!
https://www.nature.com/articles/s41588-017-0007-6
Hi,
I try to run the line "cgmaptools snv -i RR.ATCGmap.gz -m bayes -v bayes.vcf -o bayes.snv --bayes-dynamicP",
and I got an error "TypeError: unsupported operand type(s) for +: 'zip' and 'zip'"
Can you please give me guildance on how to fix it?
Thanks,
Alice
Hi Weilong,
I am Yibing, I tried to select the CDS regions using cgmaptool command as followed:
cgmaptools select -i B73_merge.CGmap -r B73_cds.bed
But the hint showed that there is a wrong parameter. Could you please tell me what's wrong? I am already made sure that region file is tab separated.
All the best,
Yibing
Dear Guo,
I want to merge multiple sample with mergelist to matrix and I use this command line to generate the index but the index is empty.
Why ? (I use python 2 with cgmaptools)
zcat 1.CGmap.gz 2.CGmap.gz 3.CGmap.gz 4.CGmap.gz | gawk '$8>=5' | cut -f1,3 | sort -u | cgmaptools sort -c 1 -p 2 > index
Waiting for your reply, and thanks a lot.
Could you give me more specific explanation about the meaning of the standard output?
Could you please give me the full name of the following items?
sense_ave_mC
sense_sum_mC
sense_sum_NO
anti_ave_mC
anti_sum_mC
anti_sum_NO
total_ave_mC
total_sum_mC
total_sum_NO
Hi,Weilong.
I want to draw a picture like Fig2.D&E in your article, CGmapTools improves the precision of heterozygous SNV calls and supports allele-specific methylation detection and visualization in bisulfite-sequencing data.
I couldn't find your related any information about tanghulu of several samples’ ASM from your website or your paper. Please give me your associative code or information about Fig2.D&E in your article.
Thanks very much.
I am looking forward to your reply.
Grace
Hi
How can I use cgmaptools for paired-end reads that aligned by bismark? At the moment cgmaptools don't support it.
Regards
Hi! I was able to run the pipeline (ATCGmap, SNV and ASM) using the test files successfully (WG.bam). I am also able to generate the ATCGmap and SNV vcf files on my own data (192 samples) successfully. However, in calling the ASM command on my data, it seems to run a long time (so far, several hours) without going to completion (it generates an empty .asm file but then stalls there). I was wondering whether you have suggestions about why this may be the case. My bam files are about 100MB, the hg19 fasta file is about 3GB and the VCF files are about 15MB. This is the pipeline I'm following:
$cd cgmaptools
$ls
bin include RICHS_test src test cgmaptools install.sh README.md
$cd RICHS_test
$ls
21007.bam 21007.bam.bai hg19.fa hg19.fa.fai
#create ATCGmap (~4min)
$../cgmaptools convert bam2cgmap -b 21007.bam -g hg19.fa --rmOverlap -o 21007
$ls
21007.ATCGmap.gz 21007.bam 21007.bam.bai hg19.fa hg19.fa.fai
#create SNV vcf file (~30min)
$../cgmaptools snv -m bayes --bayes-dynamicP -i 21007.ATCGmap.gz -v 21007.vcf
$ls
21007.vcf 21007.ATCGmap.gz 21007.bam 21007.bam.bai hg19.fa hg19.fa.fai
#create ASM file (?? hours)
$../cgmaptools asm -r hg19.fa -b 21007.bam -l 21007.vcf > 21007.asm
$ls
21007.asm (0KB) 21007.vcf (13MB) 21007.ATCGmap.gz (110MB) 21007.bam (83MB) 21007.bam.bai (3MB) hg19.fa (3GB) hg19.fa.fai (1KB)
Hi!
I've started using the toolkit and have found some DMRs and DMSs. As per the faq, you have mentioned I can intersect the outcomes using bedtools to annotate the genes.
Do I need to merge the DMR windows or DMS over a window and then annotate with the bed file?
Any suggestion is appreciated!
By looking at the coverage analysis, you mentioned the methylation-effective several times, but could you be more specific about this definition. What is "methylation-effective"? A formula would be fine.
Hi,
I'm trying to use methpat to visualize data generated with bs-seeker2 as I was used to do with bismark. The input file is 'CpG_context_xxx.txt' from bismark_methylation_extractor, and it looks like this:
Bismark methylation extractor version v0.17.0
LIN6K:00291:01397 - chr1 1160677 z
LIN6K:00291:01397 + chr1 1160714 Z
LIN6K:00291:01397 + chr1 1160728 Z
LIN6K:00291:01397 + chr1 1160730 Z
LIN6K:00291:01397 + chr1 1160736 Z
LIN6K:00291:01397 + chr1 1160761 Z
LIN6K:00291:01397 + chr1 1160804 Z
LIN6K:00291:01397 + chr1 1160819 Z
LIN6K:00291:01397 + chr1 1160827 Z
LIN6K:00291:01397 + chr1 1160876 Z
LIN6K:00291:01397 + chr1 1160884 Z
LIN6K:00291:01397 + chr1 1160886 Z
LIN6K:00291:01397 + chr1 1160904 Z
LIN6K:00291:01397 + chr1 1160924 Z
LIN6K:01879:02897 + chr1 1160714 Z
LIN6K:01879:02897 + chr1 1160728 Z
LIN6K:01879:02897 + chr1 1160730 Z
LIN6K:01879:02897 + chr1 1160736 Z
LIN6K:01879:02897 + chr1 1160761 Z
In other word, each read or fragment has multiple lines with the methylation status of each CpG. I couldn't find any output file in CGmap that would maintain the information of methylation of each molecule. Is there an easy way to extract this information from the .bam files?
Thanks !
All C-build executable returns exit(0) on printing help.
All CPP executables (ATCGmapMerge CGmapSelectByRegion CGmapMethInBed CGmapMethInFragReg) should return exit(0) as well, since calling it does what it should (print help) and then exits correctly.
This is especially helpful for testing if commands are on PATH and runs "correctly" thus exit(1) could mean another errors.
A declarative, efficient, and flexible JavaScript library for building user interfaces.
🖖 Vue.js is a progressive, incrementally-adoptable JavaScript framework for building UI on the web.
TypeScript is a superset of JavaScript that compiles to clean JavaScript output.
An Open Source Machine Learning Framework for Everyone
The Web framework for perfectionists with deadlines.
A PHP framework for web artisans
Bring data to life with SVG, Canvas and HTML. 📊📈🎉
JavaScript (JS) is a lightweight interpreted programming language with first-class functions.
Some thing interesting about web. New door for the world.
A server is a program made to process requests and deliver data to clients.
Machine learning is a way of modeling and interpreting data that allows a piece of software to respond intelligently.
Some thing interesting about visualization, use data art
Some thing interesting about game, make everyone happy.
We are working to build community through open source technology. NB: members must have two-factor auth.
Open source projects and samples from Microsoft.
Google ❤️ Open Source for everyone.
Alibaba Open Source for everyone
Data-Driven Documents codes.
China tencent open source team.