abelcarreras / dynaphopy Goto Github PK
View Code? Open in Web Editor NEWPhonon anharmonicity analysis from molecular dynamics
Home Page: http://abelcarreras.github.io/DynaPhoPy/
License: MIT License
Phonon anharmonicity analysis from molecular dynamics
Home Page: http://abelcarreras.github.io/DynaPhoPy/
License: MIT License














































































































Hi,
When I change the format of trajectory from lammpstrj to hdf5, the size is increased from 13.5 MB to 53.1 MB. It is quite wired because the file in hdf5 is expected to be smaller. Could you give me some helps?
The command for trajectory production is:
dump dynaphopy_pos all custom 100 LiSi_${t}_pos.lammpstrj x y z
dump_modify dynaphopy_pos sort id
The format transfer is conducted following:
dynaphopy input LiSi_300_pos.lammpstrj -ts 0.001 -sv LiSi_300_pos.hdf5
Hi Abel,
I have two issues.
The full power spectra become noisier when I increase the simulation length. This doesn't seem due to the concatenation though since the power spectra for simulations using dt={500ps, 1ns, 1.5ns}, all yielded the same result. I also concatenated the files using concath5, so I would assume this is done in the most consistent way with how dynaphopy works. Here, I just took the raw data from dynaphopy for the power spectra. I didn't multiply by any factor since it seemed to match the DOS, so I assumed this was already done by dynaphopy.
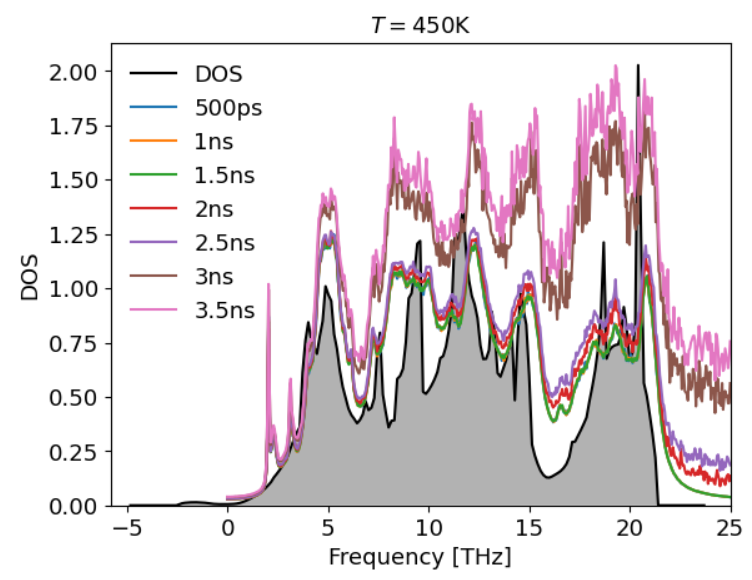
When I change the temperature, however, I see a large increase in the power spectra values
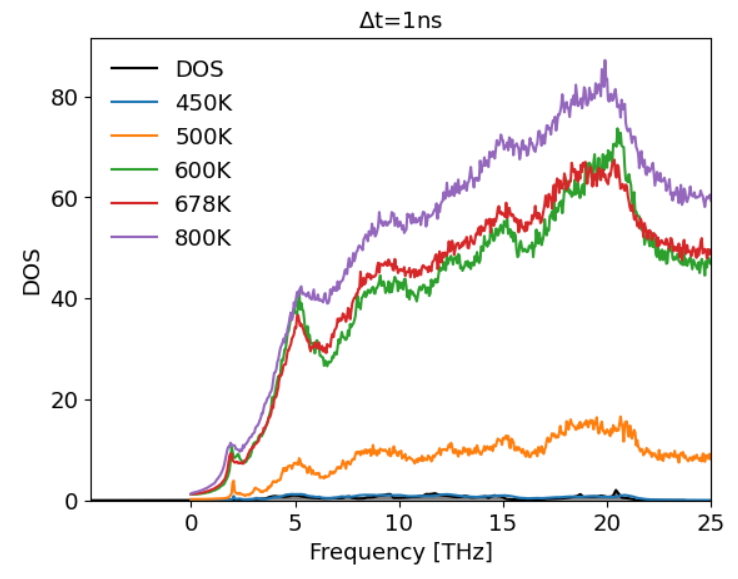
But then when I add a factor of 1/(3n_atomsk_b*T), I get odd results where high temperature spectra are kind of matching, but not lower T spectra.

I seem to get a similar problem than raised here (#17) since at higher frequency for the noisy high T spectra I get very large power spectra values in a similar way. Nevertheless, I produced my supercells using phonopy so the atoms should be rightly ordered. You mentioned there an --auto_order tag. Is there an equivalent I could use in a python script?
Many Thanks,
Gabriel
I have run MD by ASE. How to analyse it by DynaPhoPy?
Hi,
It would be great to add an hdf5 parser to dynaphopy.interface.iofile.trajectory_parsers so that we can parse hdf5 in a python script and post-process from there.
Many Thanks.
Gabriel
Hi, I'm sorry to bother you. I just started using dynaphopy, and I got the following error when running GaN _ lammps in the examples directory. What may have caused this mistake? How should I avoid such errors? The following is my error report record. Thank you for your help.
slurm-749533.txt

In the requirements line in setup.py there is a check whether the platform is windows:
install_requires=['phonopy', 'numpy', 'scipy', 'matplotlib'] + ["windows-curses"] if "win" in sys.platform else [],
On MacOS sys.platform = 'darwin' which also contains win as a substring and therefore "windows-curses" is required.
A safer check would be if sys.platform in ["win32", "cygwin"] which are the two possible options for Windows I found in the documentation of sys.platform.
Hi, I'm sorry to bother you again. I want to use phonolammps to calculate the force constants, and then use dynaphopy to further calculate the renormalized force constants. According to my understanding, the order of atoms in POSCAR for calculating force constants in phonolammps should be consistent with that in data file for calculating atomic trajectory and POSCAR file for calculating renormalized force constants in dynaphopy (I adopted amorphous SiO2 structure of 648 atoms without cell expansion). However, when I use phonolammps to calculate the force constants, the generated POSCAR file is shown in POSCAR.txt. I used this POSCAR as one of the input files of dynaphopy. When I used -c_poscar (or -c _ lammps) to generate a new vasp_poscar (data.file) file, it seemed that the type of atoms could not be completely recognized correctly.
When using lammps to calculate the atomic trajectory, I can manually change the atomic type in data.file through some simple post-processing to make it consistent with the POSCAR file generated by phonolammps. Can I directly use this POSCAR generated by phonolammps as the input file when calculating the renormalized force constants and linewidth by dynaphopy? Because dynaphopy's -c_lammps and -c_poscar commands don't correctly identify this POSCAR file, I'm not sure whether it can correctly identify this POSCAR file when calculating the renormalized force constants.
I wish I had made my question clear. Thank you very much for your help.
POSCAR.txt
data.file.txt
vasp_posacr.txt
Dear Abel Carreras:
Hello, I have a question related to vibrational free energy calculated by DynaPhopy.
I want to calculate the vibrational free energy of amorphous by considering anharmonicity, so I'm performing VASP MD in various temperatures. The values of quasi-particle and power spectrum free energy are quite similar to the harmonic free energy. Thus, the anharmonic free energy value does not seem unreasonable.
However, can DynaPhopy be applied to the non-crystalline system like the amorphous phase?
Kun Hee Ye
Since 1.17.0, there is no tag tag on GitHub, Can you tag on github when pip releases a new version?
When I package for ArchLinux on Aur: python-dynaphopy, I can't get the latest source code, currently only on pip, pip's download connection is not fixed, it is very inconvenient when packing
Dear Abel Carreras:
I have tried to repeat your example to calculate the renormalized phonon dispersion but there are some bugs I can't solve. The output picture is blank as follows and all the commands are input as your README file. I also use the Si to test, the same blank picture is got too. Could you please help me solve this problem?

Hi Abel,
I noticed that I had to adjust the symmetry tolerance from 1e-5 to 1e-2 to ensure that Phonopy recognized the space group correctly for my system.
I am also hoping to try out force constant symmetrization using DynaPhoPy. I have noticed that the symprec parameter has a default value of 1e-5 when flag --fcsymm is used. Is there a way to easily modify symprec?
Best,
Bob
Hello, I have encountered some problems when I calculate phonon free energy with dynaphopy, hope to get your help.
The harmonic phonon free energy calculated by dynaphopy and phonopy are very different, but both calculations use the same FORCE_SETS. I want to know whether dynaphopy calculates the harmonic phonon properties by calling phonopy? If so, why do I get the two results are different? This makes me can 't believe the result of the anharmonic phonon free energy calculated by dynaphopy.
I use the --thm_full tags to calculates the quasiparticle phonon free energy and the power spectrum phonon free energy. However, these results also are different, i also use the renormalized force constants to calculates anharmonic phonon free energy with phonopy, but this result is different from the previous two. Besides, By examining the mesh.yaml file generated by the phonopy calculation, I found that the phonon frequencies calculated using the renormalized force constant have eight zero frequencies in the Brillouin zone ( gamma point ). I want to know whether these situations means that my MD trajectory is not appropriate? My MD trajectory consist of 50000 time steps, relaxation of 500 ps before sampling, my time step is 0.001ps. Do I need to perform a longer simulation and sample a larger MD trajectory(100000 time steps)? By the way, in order to get a more accurate result, how much MD trajectory does dynaphopy usually need?
Dear Abel carreras:
We need to calculate the spectral function of the material to give the information of different components. Can we solve this problem with your software? Additionally, the picture as follows is the 4 - Frequency vs linewidth (interpolated mesh) of GaN. Can you please explain what the different colors represent?

Hi Abel,
I found the link to the Anaconda-supported installation of DynaPhoPy https://anaconda.org/conda-forge/dynaphopy. It provides an alternate way conda install -c conda-forge dynaphopy to install DynaPhonPy,
However, neither the GitHub nor the DynaPhoPy website mentioned via conda install method.
The ways that I have seen are
pip install dynaphopy --user or python setup.py install --user from GitHub,
python setup.py install --user or python setup_openmp.py install --user from DynaPhoPy.
I was wondering if the conda install method is recommended and who maintains it.
Best regards,
Huan
Hello,
I want to use DynaPhoPy to analyse three 100ps runs generated with the following INCAR on VASP:

The issue is that I don't think DynaPhoPy reads my XDATCAR knowing that NBLOCK = 50 and therefore interprets it as a regular XDATCAR with about 1,000 MD steps, while there are actually about 50,000 steps. I say about because I have cat together 7 runs due to walltime limits and there might be a few steps missing here and there.
Another point that raised concern is that even when I set my timestep to a particular value, so in this case -ts 0.002, DynaPhoPy states another value - see screenshot of the interactive interface after entering the command dynaphopy input_file 300_XDATCAR -ts 0.002 -i:

This may be due to my NBLOCK setting again because I tried setting various timestep using -ts on a short simulation with NBLOCK = 1 and DynaPhoPy worked fine - see screenshot of the interactive interface after entering the command dynaphopy input_file XDATCAR -ts 0.00005 -i when the timestep actually used was 1fs:

Looking at the source code, there is only one mention of NBLOCK but in the unittest and it looks like that you assume it is 1 since in the line time = np.array([i*time_step for i in range(trajectory.shape[0])], dtype=float) you assume that the total simulation time is timestep*number_of_steps.
Kind Regards,
Gabriel
Dear dynaphopy team,
sorry to bother you with this issue. I try to run the dynaphopy to obtain the phonon frequency and linewidths. However, when I run the command like below:
dynaphopy input_file XDATCAR -ts 0.02 -sdata
there is a confused bug:
Reading VASP POSCAR structure
Reading XDATCAR file
This could take long, please wait..
No velocity provided! calculating it from coordinates...
MD cell size relation: [2 2 2]
Using 20000 steps
Traceback (most recent call last):
File "/home/zeng/.local/bin/dynaphopy", line 4, in
import('pkg_resources').run_script('dynaphopy==1.16.6', 'dynaphopy')
File "/home/zeng/.local/lib/python2.7/site-packages/pkg_resources/init.py", line 666, in run_script
self.require(requires)[0].run_script(script_name, ns)
File "/home/zeng/.local/lib/python2.7/site-packages/pkg_resources/init.py", line 1469, in run_script
exec(script_code, namespace, namespace)
File "/home/zeng/.local/lib/python2.7/site-packages/dynaphopy-1.16.6-py2.7-linux-x86_64.egg/EGG-INFO/scripts/dynaphopy", line 362, in
File "build/bdist.linux-x86_64/egg/dynaphopy/init.py", line 1196, in write_quasiparticles_data
File "build/bdist.linux-x86_64/egg/dynaphopy/init.py", line 1086, in get_commensurate_points_data
File "build/bdist.linux-x86_64/egg/dynaphopy/interface/phonopy_link.py", line 155, in obtain_phonopy_dos
File "build/bdist.linux-x86_64/egg/dynaphopy/interface/phonopy_link.py", line 79, in get_phonon
File "build/bdist.linux-x86_64/egg/dynaphopy/atoms.py", line 97, in get_supercell_phonon
File "build/bdist.linux-x86_64/egg/dynaphopy/atoms.py", line 187, in get_force_sets
TypeError: 'NoneType' object has no attribute 'getitem'
I try to obtain the temperature profile and it is worked. I also test the examples and worked very well.
Could you give me some advice about this problem?
Dear Abel,
I have successfully compiled phonopy-2.14.0 and then dynaphopy-1.17.15. I am certain that the path 'phonopy-2.14.0/lib/python' was added to PYTHONPATH after compiling Phonopy. However, I am still encountering this issue when running dynaphopy. By the way, running phonolammps is everything fine.
File "/home/dynaphopy-1.17.15/lib64/python3.6/site-packages/dynaphopy-1.17.15-py3.6-linux-x86_64.egg/dynaphopy/interface/phonopy_link.py", line 2, in
from phonopy.api_phonopy import Phonopy
ModuleNotFoundError: No module named 'phonopy'
Thanks,
Te-Huan
Hi, professor.
I would like to use the dynaphopy by openmp, but when I reinstall the dynaphopy by command like "python setup_openmp.py install --user", I cannot find the setup_openmp.py file and there is only setup.py file.
I tried to load the python/3.7.0 and gcc/6.3.0 in the supercomputer system and then used the command:
"python setup.py install --user" to install.
The installation is successful and I found the prompt information indicates that the openmp is used to install.
But when I used the command like:
mpirun -np 36 dynaphopy input_file dump.traj -sdata
to run the dynaphopy, I found there is no reaction to my command. But if I used:
dynaphopy input_file dump.traj -sdata
it can run slowly.
Could you tell me how to run the dynaphopy by openmp (multiple cores)?
Best,
zezhu
If I run the test script Si_test.py I get an error
ValueError: operands could not be broadcast together with shapes in the test test_force_constants_self_consistency.
def test_force_constants_self_consistency(self):
self.calculation.select_power_spectra_algorithm(2)
renormalized_force_constants = self.calculation.get_renormalized_force_constants().get_array()
harmonic_force_constants = self.calculation.dynamic.structure.get_force_constants().get_array()
self.assertEqual(np.allclose(renormalized_force_constants, harmonic_force_constants, rtol=1, atol=1.e-2), True)The issue appears to be that renormalized_force_constants and harmonic_force_constants have different shapes. In this case these array shapes are (2,64,3,3) and (64,64,3,3), so np.allclose() raises a ValueError.
Hi Abel,
I have been doing a few tests on DynaPhoPy with my data but my results do not make sense, I think NBLOCK might be the problem. Just a summary of the outcome of our last exchange, I re-wrote my XDATCAR in a sequential way, from 1 to 996 in steps of 1, even though my NBLOCK is 50, and we concluded that this file with a timestep_per_block = timestep*NBLOCK should work.
But first of all, when I run the Maxwell-Boltzmann Distribution Analysis, it gives me a temperature fit of 6.6K, but I had a quick glance at my OSZICAR files and the temperaturestays around 300K, and for sure it does not settle down at 6.6K.

But I anticipate this is due to my use of NBLOCK since this calculation is dependent on the timestep and my timestep is the timestep per block, which is significantly larger but atoms are likely to move periodically and therefore from structure to structure it is likely that there isn't much change, hence reducing the fitted temperature of the system.
I therefore run dynaphopy on a previous run on 32 atoms I did with a timestep of 1fs for 1,000 steps with NBLOCK=1, and this time I got a much more sensible temperature, 287.8K.

The more worrying thing about this is that I wonder if DynaPhoPy is therefore also having trouble interpreting the velocities (since it calculates it from the coordinates of the trajectory), which is at the core of your model and of your technique to calculate the power spectra. So, I wonder if NBLOCK could screw the whole calculations basically.
What reinforces this idea is that the Maxwell Boltzmann Distribution Analysis is not the only feature misbehaving. I have run a peak analysis of the phonon projected power spectrum, and I also get weird results. Here is a snapshot of the output for one mode and its corresponding plot, but in summary


But not even going into that much details, the power spectrum projected onto the gamma point is also not looking good, as I expect it to give me peaks around the frequency of each of the 12 modes present, and I only get 3 here...

I tried the peak analysis on my previous run and it looks somewhat better, at least for the fitting error, even though the frequency still looks wrong.


But I suspect this is mainly due to the fact that I only use 0.85ps worth of simulation and 32 atoms, which means I also use much less coefficients for the algorithm used to compute the power spectra from the velocities since there is a requirement that nb_of_step>nb_of_coeff, even though I cannot usew that many for the other simulations either. But in my opinion, it is quite striking to see that the fitting error is smaller for my 0.85ps run with 32 atoms than for my 100ps run with 256 atoms... So in my opinion there imight be an issue in the code interpretation of NBLOCK and maybe it simply cannot work with an NBLOCK different than 1. But I might doing another mistakes.
Let me know your thoughts on this,
Gabriel
Dear Abel Carreras
I hope you are doing well. I have run into an issue when I was trying to compute the full power spectrum for a larger MD supercell. When I have calculated the full power spectrum for 1 1 4 supercell (120 atoms), I have found a good match with the harmonic dos (of course 1 1 4 supercell as well). But, When I increased the supercell to 2 2 4 (480 atoms), I have found that the high frequency regime of the power spectrum does not match with the harmonic phonon dos for 2 2 4 supercell although there is some similarity. I have attached the two plots here for your reference. Please let me know your thoughts on this. For your information, I have used 30 ps of trajectories (1 fs timestep) to calculate the power spectrum for both supercell. Therefore, I am thinking the length of the simulation may not be an issue. Thank you very much for your time.


Best
Ballal
Hello,
I have started recently using this code to calculate phonon properties from MD simulations. I am using LAMMPS software to perform MD simulation. I get a trajectory of atomic motion for fairly large cell (2k atoms) for 100 k timesteps with 1fs timestep.
I calculate FORCE SETS using phonopy for 8 times smaller cell (phonopy cell is 5x5x5 and MD cell is 10x10x10). When I try calculating phonon band structure with phonopy using these force sets I get reasonably good agreement with DFT. If I try to calculate power spectra projected on phonon modes using Dynaphopy I get following error:
Traceback (most recent call last):
File "/sfihome/djordje.dangic/anaconda2/envs/phonopy/lib/python3.7/site-packages/phonopy/api_phonopy.py", line 2951, in _build_primitive_cell
self._supercell, trans_mat, self._symprec)
File "/sfihome/djordje.dangic/anaconda2/envs/phonopy/lib/python3.7/site-packages/phonopy/structure/cells.py", line 49, in get_primitive
return Primitive(supercell, primitive_frame, symprec=symprec)
File "/sfihome/djordje.dangic/anaconda2/envs/phonopy/lib/python3.7/site-packages/phonopy/structure/cells.py", line 356, in init
self._primitive_cell(supercell)
File "/sfihome/djordje.dangic/anaconda2/envs/phonopy/lib/python3.7/site-packages/phonopy/structure/cells.py", line 415, in _primitive_cell
raise ValueError
ValueError
During handling of the above exception, another exception occurred:
Traceback (most recent call last):
File "/sfihome/djordje.dangic/anaconda2/envs/phonopy/bin/dynaphopy", line 343, in
calculation.write_power_spectrum_phonon(args.save_phonon_mode[0])
File "/sfihome/djordje.dangic/anaconda2/envs/phonopy/lib/python3.7/site-packages/dynaphopy/init.py", line 1030, in write_power_spectrum_phonon
self.get_power_spectrum_phonon(),
File "/sfihome/djordje.dangic/anaconda2/envs/phonopy/lib/python3.7/site-packages/dynaphopy/init.py", line 716, in get_power_spectrum_phonon
(power_spectrum_functions[self.parameters.power_spectra_algorithm])[0](self.get_vq(),
File "/sfihome/djordje.dangic/anaconda2/envs/phonopy/lib/python3.7/site-packages/dynaphopy/init.py", line 632, in get_vq
self._vq = projection.project_onto_phonon(self.get_vc(), self.get_eigenvectors())
File "/sfihome/djordje.dangic/anaconda2/envs/phonopy/lib/python3.7/site-packages/dynaphopy/init.py", line 169, in get_eigenvectors
self.parameters.reduced_q_vector))
File "/sfihome/djordje.dangic/anaconda2/envs/phonopy/lib/python3.7/site-packages/dynaphopy/interface/phonopy_link.py", line 117, in obtain_eigenvectors_and_frequencies
phonon = get_phonon(structure)
File "/sfihome/djordje.dangic/anaconda2/envs/phonopy/lib/python3.7/site-packages/dynaphopy/interface/phonopy_link.py", line 90, in get_phonon
symprec=1e-5)
File "/sfihome/djordje.dangic/anaconda2/envs/phonopy/lib/python3.7/site-packages/phonopy/api_phonopy.py", line 117, in init
self._build_primitive_cell()
File "/sfihome/djordje.dangic/anaconda2/envs/phonopy/lib/python3.7/site-packages/phonopy/api_phonopy.py", line 2955, in _build_primitive_cell
raise RuntimeError(msg)
RuntimeError: Creating primitive cell is failed. PRIMITIVE_AXIS may be incorrectly specified.
Do you have any suggestions how to avoid this. Calculating DOS with MD works fine, as well as calculating power spectra (PS) projected on reciprocal lattice vector although results are a bit strange. I get phonons where I should not have any and the PS does not fall to zero for frequencies larger than Debye frequency from phonopy.
Thanks in advance,
Djordje
Hi,
I want to confirm whether the dynaphopy can be applied to calculate the phonon lifetime vs frequency as attached figure?
Thanks,

Dear Abel Carreras,
I installed the dynaphopy package and tested the Si example you gave in the doc. VASP was used for the testing, with 1E4 MD steps at 800 K. Then I ran the dynaphopy script in interactive mode, using the OUTCAR produced by VASP calculation. The Maxwell-Boltzmann distribution analysis plotted a graph and printed a fitting temperature (2022.4 K), but I found the fitting curve deviated too much from the data, as shown below.

I tried to change the number of MD steps used for analysis by specifying -n parameter, i.e. -n 8000 or -n 5000, resulting in exactly the same fitting results. It seems it did not really fit the data at all.
More Information on my setup:
A declarative, efficient, and flexible JavaScript library for building user interfaces.
🖖 Vue.js is a progressive, incrementally-adoptable JavaScript framework for building UI on the web.
TypeScript is a superset of JavaScript that compiles to clean JavaScript output.
An Open Source Machine Learning Framework for Everyone
The Web framework for perfectionists with deadlines.
A PHP framework for web artisans
Bring data to life with SVG, Canvas and HTML. 📊📈🎉
JavaScript (JS) is a lightweight interpreted programming language with first-class functions.
Some thing interesting about web. New door for the world.
A server is a program made to process requests and deliver data to clients.
Machine learning is a way of modeling and interpreting data that allows a piece of software to respond intelligently.
Some thing interesting about visualization, use data art
Some thing interesting about game, make everyone happy.
We are working to build community through open source technology. NB: members must have two-factor auth.
Open source projects and samples from Microsoft.
Google ❤️ Open Source for everyone.
Alibaba Open Source for everyone
Data-Driven Documents codes.
China tencent open source team.