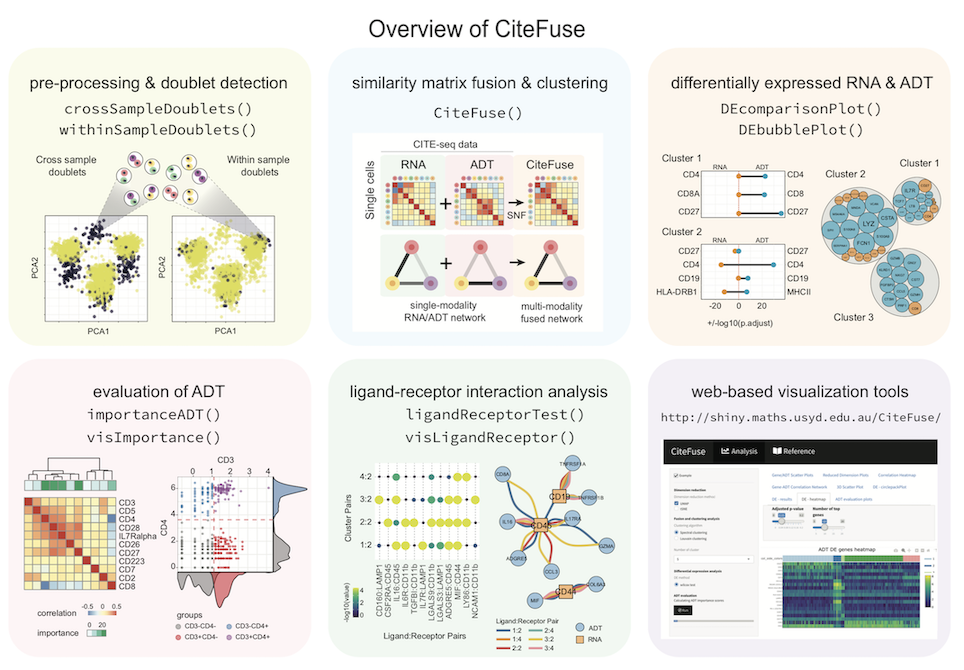
CiteFuse is a streamlined package consisting of a suite of tools for pre-processing, modality integration, clustering, differential RNA and ADT expression analysis, ADT evaluation, ligand-receptor interaction analysis, and interactive web-based visualization of CITE-seq data
Install CiteFuse via Bioconductor
library(BiocManager)
BiocManager::install("CiteFuse")Install github version CiteFuse
BiocManager::install("SydneyBioX/CiteFuse")You can find the vignette at our website: https://sydneybiox.github.io/CiteFuse/articles/CiteFuse.html.
Hani Jieun Kim, Yingxin Lin, Thomas A Geddes, Jean Yee Hwa Yang, Pengyi Yang, CiteFuse enables multi-modal analysis of CITE-seq data, Bioinformatics, Volume 36, Issue 14, 15 July 2020, Pages 4137–4143, https://doi.org/10.1093/bioinformatics/btaa282





































