single-cell-genetics / mquad Goto Github PK
View Code? Open in Web Editor NEWTool for modelling of mitochondrial heteroplasmy
Home Page: https://github.com/aaronkwc/MQuad_paper_reproduced_results
License: Apache License 2.0
Tool for modelling of mitochondrial heteroplasmy
Home Page: https://github.com/aaronkwc/MQuad_paper_reproduced_results
License: Apache License 2.0




















Hi.
I had a question
which section of your scripts made the heat map? my mquad had a bug when I ran it where it did not plot the heat maps in a format which is visible and I dont want to rerun everything again. Id ideally just utilise my outputs to make the heatmap myself
Thank you !
deltaBIC_cdf.pdf
top variants heatmap.pdf
With reference to the two outputs above, in 1 out of 3 of my datasets, I am unable to identify mitochondrial variants. Manually setting the cutoff to 0.9 results in more variants being identified, but there is no diversity among the cells, despite this being a tumour sample where a mutational burden is expected (and does occur in the 2 other samples)
Any advice on can give on this is greatly appreciated. The MQuad inputs were pre-processed with cellSNP lite, using 10X cell ranger's possorted bam and barcodes, with --chrom specific to chrM
Hi,
Could I use MQuad for the whole genome scATAC data analysis and not only for chrMt?
I used the following command :
mquad -o Y_F1/ --minDP 5 --vcfData Y_F1/Y_F1_SNP/cellSNP.cells.vcf.gz
and the error is below:
File "/jdfsbjcas1/ST_BJ/PUB/User/kangjingmin/Pipline/local/bin/mquad", line 8, in
sys.exit(main())
File "/home/kangjingmin/.local/lib/python3.9/site-packages/mquad/mquad_CLI.py", line 132, in main
df = mdphd.fit_deltaBIC(
File "/home/kangjingmin/.local/lib/python3.9/site-packages/mquad/mquad_batch_mixbin.py", line 132, in fit_deltaBIC
x, y = zip(*[(r, c) for r, c in zip(dp_row, dp_col) if r in valid_rows])
ValueError: not enough values to unpack (expected 2, got 0)
Could you help me!
Thank you!
Hi. I am running mquad with 10X scRNA seq data using the following command:
mquad -m cellSNP.tag.AD.mtx, cellSNP.tag.DP.mtx -o $output_dir --minDP 5
and I received this in my log while running it:
_Fitting in sparse mode...
Initializing fit(mode: deltaBIC) on 16569 variants...
mquad_env/lib/python3.9/site-packages/bbmix/models/binomial_mix_sparse.py:156: RuntimeWarning: divide by zero encountered in log + y * np.log(p) + (n - y) * np.log(1 - p) + np.log(pi)
mquad_env/lib/python3.9/site-packages/bbmix/models/binomial_mix_sparse.py:156: RuntimeWarning: invalid value encountered in multiply + y * np.log(p) + (n - y) * np.log(1 - p) + np.log(pi)
mquad_env/lib/python3.9/site-packages/bbmix/models/binomial_mix_sparse.py:116: RuntimeWarning: divide by zero encountered in lognp.log(1 - p_k) + np.log(pi_k)
mquad_env/lib/python3.9/site-packages/bbmix/models/binomial_mix_sparse.py:115: RuntimeWarning: invalid value encountered in multiply E_gammas[k] = y * np.log(p_k) + (n - y) * _
I used --min DP5 as it is recommended for 10X data according to the manual. Do you happen to know why this issue is there or what this could mean? it appears that the package dependencies aren't correlating like they should but I don't fully understand this.
Thank you in advance!
Hi,
I try to run MQuad on 10x scRNA-seq, parameters are set like this:
mquad --c $cellSNP_lite_output -o $wkdir/Pt_12_Dx_MQuad/mquad \
-p 5 --batchFit 1 --batchSize 5 --minDP 5
The passed_variant_names.txt contains only 3 variants.
I've tried to use a lower --minDP, but it doesn't help.
What should I do to get more output variants?
Thank you in advance.
I was trying to combine my MQuad outputs with vireo. I was wondering which MQuad outputs are passed to which arguments of vireo. Thanks!
P.S. The tutorial here does not load for me:
https://github.com/single-cell-genetics/vireo/blob/master/examples/vireoSNP_clones.ipynb
After running cellSNP on my 10X scRNA bam with parameters -p 22 --minMAF 0.1 --minCOUNT 100 --chrom chrMT I get the assumed correct output files [Attached]
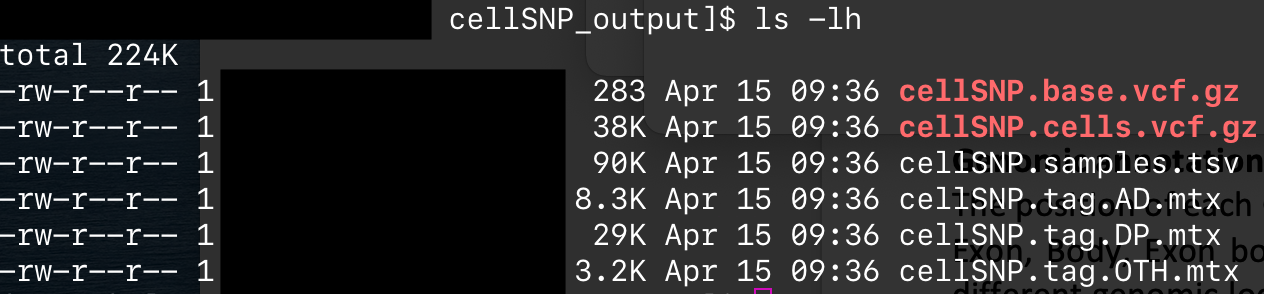
but when I run MQuad on the cellSNP output using parameters -p 20 --minDP 5 I get ValueError: cannot reshape array of size 0 into shape (0,newaxis). I've run this on 2 separate datasets and receive the same error. Could this be a python version issue? I'm on Python v.3.9.2.
Hi!
I would like to reflect the mitochondrial genetic mutation results obtained by MQuad in the Dimplot created in Seurat (like Fig. 5d in your article about MQuad).
So, I need to link AD/DP matrices and cell barcodes.
Do the 1st, 2nd and 3rd columns of "passed_ad/dp.mtx" mean variants, cell barcodes and depth(ad/dp) respectively?
And the numbers of variants and cell barcodes accord with the order of "passed_variant_names.txt" and "cellSNP.samples.tsv"(which is in the input directory)?
Hi
I notice in your paper (https://www.nature.com/articles/s41467-022-28845-0) you use SCITE to approximate lineage of cells.
Can I perhaps see the code you used (with the parameters) for running SCITE?
I understand it would look something like this:
../infSCITE
-i MutationMatrix.csv
-n 23
-m 10
-r 1
-l 700000
-fd 3.45e-3
-ad 1.46e-1
-s
-e .2
-p 10000
-d -rec 13
Alan
Traceback (most recent call last):
_ad = np.squeeze(np.asarray(self.ad[_x, _y]))
TypeError: 'coo_matrix' object is not subscriptable
Hi,
I'm trying to fit mquad on the output of cellsnp for 3' 10x single cell data. I got the following error:
16569 variants detected...
Variant names detected...
CPUs used: 20, batch size: 5
Fitting in sparse mode...
Initializing fit(mode: deltaBIC) on 16569 variants...
/Users/nrohani/mquad_env1/lib/python3.10/site-packages/bbmix/models/binomial_mix_sparse.py:156: RuntimeWarning: divide by zero encountered in log
/Users/nrohani/mquad_env1/lib/python3.10/site-packages/bbmix/models/binomial_mix_sparse.py:156: RuntimeWarning: invalid value encountered in multiply
/Users/nrohani/mquad_env1/lib/python3.10/site-packages/bbmix/models/binomial_mix_sparse.py:116: RuntimeWarning: divide by zero encountered in log
np.log(1 - p_k) + np.log(pi_k)
/Users/nrohani/mquad_env1/lib/python3.10/site-packages/bbmix/models/binomial_mix_sparse.py:115: RuntimeWarning: invalid value encountered in multiply
E_gammas[k] = y * np.log(p_k) + (n - y) * \
/Users/nrohani/mquad_env1/lib/python3.10/site-packages/bbmix/models/binomial_mix_sparse.py:156: RuntimeWarning: divide by zero encountered in log
/Users/nrohani/mquad_env1/lib/python3.10/site-packages/bbmix/models/binomial_mix_sparse.py:156: RuntimeWarning: invalid value encountered in multiply
/Users/nrohani/mquad_env1/lib/python3.10/site-packages/bbmix/models/binomial_mix_sparse.py:116: RuntimeWarning: divide by zero encountered in log
np.log(1 - p_k) + np.log(pi_k)
/Users/nrohani/mquad_env1/lib/python3.10/site-packages/bbmix/models/binomial_mix_sparse.py:115: RuntimeWarning: invalid value encountered in multiply
E_gammas[k] = y * np.log(p_k) + (n - y) * \
/Users/nrohani/mquad_env1/lib/python3.10/site-packages/bbmix/models/binomial_mix_sparse.py:156: RuntimeWarning: divide by zero encountered in log
/Users/nrohani/mquad_env1/lib/python3.10/site-packages/bbmix/models/binomial_mix_sparse.py:156: RuntimeWarning: invalid value encountered in multiply
/Users/nrohani/mquad_env1/lib/python3.10/site-packages/bbmix/models/binomial_mix_sparse.py:116: RuntimeWarning: divide by zero encountered in log
np.log(1 - p_k) + np.log(pi_k)
/Users/nrohani/mquad_env1/lib/python3.10/site-packages/bbmix/models/binomial_mix_sparse.py:115: RuntimeWarning: invalid value encountered in multiply
E_gammas[k] = y * np.log(p_k) + (n - y) * \
/Users/nrohani/mquad_env1/lib/python3.10/site-packages/bbmix/models/binomial_mix_sparse.py:156: RuntimeWarning: divide by zero encountered in log
/Users/nrohani/mquad_env1/lib/python3.10/site-packages/bbmix/models/binomial_mix_sparse.py:156: RuntimeWarning: invalid value encountered in multiply
/Users/nrohani/mquad_env1/lib/python3.10/site-packages/bbmix/models/binomial_mix_sparse.py:116: RuntimeWarning: divide by zero encountered in log
np.log(1 - p_k) + np.log(pi_k)
/Users/nrohani/mquad_env1/lib/python3.10/site-packages/bbmix/models/binomial_mix_sparse.py:115: RuntimeWarning: invalid value encountered in multiply
E_gammas[k] = y * np.log(p_k) + (n - y) * \
/Users/nrohani/mquad_env1/lib/python3.10/site-packages/bbmix/models/binomial_mix_sparse.py:156: RuntimeWarning: divide by zero encountered in log
/Users/nrohani/mquad_env1/lib/python3.10/site-packages/bbmix/models/binomial_mix_sparse.py:156: RuntimeWarning: invalid value encountered in multiply
/Users/nrohani/mquad_env1/lib/python3.10/site-packages/bbmix/models/binomial_mix_sparse.py:116: RuntimeWarning: divide by zero encountered in log
np.log(1 - p_k) + np.log(pi_k)
/Users/nrohani/mquad_env1/lib/python3.10/site-packages/bbmix/models/binomial_mix_sparse.py:115: RuntimeWarning: invalid value encountered in multiply
E_gammas[k] = y * np.log(p_k) + (n - y) * \
/Users/nrohani/mquad_env1/lib/python3.10/site-packages/bbmix/models/binomial_mix_sparse.py:156: RuntimeWarning: divide by zero encountered in log
/Users/nrohani/mquad_env1/lib/python3.10/site-packages/bbmix/models/binomial_mix_sparse.py:156: RuntimeWarning: invalid value encountered in multiply
/Users/nrohani/mquad_env1/lib/python3.10/site-packages/bbmix/models/binomial_mix_sparse.py:116: RuntimeWarning: divide by zero encountered in log
np.log(1 - p_k) + np.log(pi_k)
/Users/nrohani/mquad_env1/lib/python3.10/site-packages/bbmix/models/binomial_mix_sparse.py:115: RuntimeWarning: invalid value encountered in multiply
E_gammas[k] = y * np.log(p_k) + (n - y) * \
/Users/nrohani/mquad_env1/lib/python3.10/site-packages/bbmix/models/binomial_mix_sparse.py:156: RuntimeWarning: divide by zero encountered in log
/Users/nrohani/mquad_env1/lib/python3.10/site-packages/bbmix/models/binomial_mix_sparse.py:156: RuntimeWarning: invalid value encountered in multiply
/Users/nrohani/mquad_env1/lib/python3.10/site-packages/bbmix/models/binomial_mix_sparse.py:116: RuntimeWarning: divide by zero encountered in log
np.log(1 - p_k) + np.log(pi_k)
/Users/nrohani/mquad_env1/lib/python3.10/site-packages/bbmix/models/binomial_mix_sparse.py:115: RuntimeWarning: invalid value encountered in multiply
E_gammas[k] = y * np.log(p_k) + (n - y) * \
/Users/nrohani/mquad_env1/lib/python3.10/site-packages/bbmix/models/binomial_mix_sparse.py:156: RuntimeWarning: divide by zero encountered in log
/Users/nrohani/mquad_env1/lib/python3.10/site-packages/bbmix/models/binomial_mix_sparse.py:156: RuntimeWarning: invalid value encountered in multiply
/Users/nrohani/mquad_env1/lib/python3.10/site-packages/bbmix/models/binomial_mix_sparse.py:116: RuntimeWarning: divide by zero encountered in log
np.log(1 - p_k) + np.log(pi_k)
/Users/nrohani/mquad_env1/lib/python3.10/site-packages/bbmix/models/binomial_mix_sparse.py:115: RuntimeWarning: invalid value encountered in multiply
E_gammas[k] = y * np.log(p_k) + (n - y) * \
/Users/nrohani/mquad_env1/lib/python3.10/site-packages/bbmix/models/binomial_mix_sparse.py:156: RuntimeWarning: divide by zero encountered in log
/Users/nrohani/mquad_env1/lib/python3.10/site-packages/bbmix/models/binomial_mix_sparse.py:156: RuntimeWarning: invalid value encountered in multiply
/Users/nrohani/mquad_env1/lib/python3.10/site-packages/bbmix/models/binomial_mix_sparse.py:116: RuntimeWarning: divide by zero encountered in log
np.log(1 - p_k) + np.log(pi_k)
/Users/nrohani/mquad_env1/lib/python3.10/site-packages/bbmix/models/binomial_mix_sparse.py:115: RuntimeWarning: invalid value encountered in multiply
E_gammas[k] = y * np.log(p_k) + (n - y) * \
deltaBIC was calculated for 16569 variants and took:46.29 minutes
Out directory already exists, overwriting content inside...
deltaBIC cutoff = 666.1358156537099
Number of variants passing threshold: 91
Traceback (most recent call last):
File "/Users/nrohani/mquad_env1/bin/mquad", line 8, in
sys.exit(main())
File "/Users/nrohani/mquad_env1/lib/python3.10/site-packages/mquad/mquad_CLI.py", line 144, in main
best_ad, best_dp = mdphd.selectInformativeVariants(min_cells = minCell, out_dir = out_dir, tenx_cutoff=cutoff)
File "/Users/nrohani/mquad_env1/lib/python3.10/site-packages/mquad/mquad_batch_mixbin.py", line 231, in selectInformativeVariants
renamed_vars.append((var.split('_')[1] + var.split('_')[2] + '>' + var.split('_')[3]))
IndexError: list index out of range
am encountering the same issue that was noticed by another user.
my input is two matrices, AD and DP
mquad -m cellSNP.tag.AD.mtx,cellSNP.tag.DP.mtx -o ./mquad/ --minDP 5 -p 20
output
[MQuad] Loading matrix files ...
16569 variants detected...
Variant names detected...
CPUs used: 20, batch size: 128
Fitting in sparse mode...
Initializing fit(mode: deltaBIC) on 16569 variants...
Traceback (most recent call last):
File "/Users/nikolay/anaconda3/envs/python-3.11.3/bin/mquad", line 8, in
sys.exit(main())
^^^^^^
File "/Users/nikolay/anaconda3/envs/Python-3.11.3/lib/python3.11/site-packages/mquad/mquad_CLI.py", line 132, in main
df = mdphd.fit_deltaBIC(
^^^^^^^^^^^^^^^^^^^
File "/Users/nikolay/anaconda3/envs/Python-3.11.3/lib/python3.11/site-packages/mquad/mquad_batch_mixbin.py", line 142, in fit_deltaBIC
results = pool.starmap_async(fit_batch, self._batchify(batch_size, x, y, valid_row_sizes)).get()
^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^
File "/Users/nikolay/anaconda3/envs/Python-3.11.3/lib/python3.11/multiprocessing/pool.py", line 382, in starmap_async
return self._map_async(func, iterable, starmapstar, chunksize,
^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^
File "/Users/nikolay/anaconda3/envs/Python-3.11.3/lib/python3.11/multiprocessing/pool.py", line 478, in _map_async
iterable = list(iterable)
^^^^^^^^^^^^^^
File "/Users/nikolay/anaconda3/envs/Python-3.11.3/lib/python3.11/site-packages/mquad/mquad_batch_mixbin.py", line 80, in _batchify
_ad = np.squeeze(np.asarray(self.ad[_x, _y]))
TypeError: 'coo_matrix' object is not subscriptable
Hi!
disclaimer: I am not familiar with genotyping.
I was thinking about using MQuad on my scRNAseq data. The guide says I should use cellSNP/cellSNP-lite first. However, these would detect variants in the entire genome, not just mitochondrial ones. Should I subset the original cellranger bam file to only contain the mito reads?
Hi all,
I met dependency conflicts when installing MQuad.
Installing collected packages: sklearn, pytz, zipp, tzdata, six, pyparsing, pillow, packaging, numpy, kiwisolver, fonttools, cycler, scipy, python-dateutil, importlib-resources, contourpy, pandas, matplotlib, kneed, vireoSNP, BBMix, mquad
ERROR: pip's dependency resolver does not currently take into account all the packages that are installed. This behaviour is the source of the following dependency conflicts.
dynamo-release 1.3.3 requires PATSY>=0.5.1, which is not installed.
dynamo-release 1.3.3 requires scikit-learn>=0.19.1, which is not installed.
dynamo-release 1.3.3 requires statsmodels>=0.9.0, which is not installed.
dynamo-release 1.3.3 requires tqdm, which is not installed.
dynamo-release 1.3.3 requires typing-extensions, which is not installed.
pynndescent 0.5.10 requires joblib>=0.11, which is not installed.
pynndescent 0.5.10 requires scikit-learn>=0.18, which is not installed.
umap-learn 0.5.3 requires scikit-learn>=0.22, which is not installed.
umap-learn 0.5.3 requires tqdm, which is not installed.
Successfully installed BBMix-0.2.1 contourpy-1.1.1 cycler-0.12.0 fonttools-4.42.1 importlib-resources-6.1.0 kiwisolver-1.4.5 kneed-0.8.5 matplotlib-3.7.3 mquad-0.1.7 numpy-1.24.4 packaging-23.1 pandas-2.0.3 pillow-10.0.1 pyparsing-3.1.1 python-dateutil-2.8.2 pytz-2023.3.post1 scipy-1.10.1 six-1.16.0 sklearn-0.0.post9 tzdata-2023.3 vireoSNP-0.5.8 zipp-3.17.0
I've tried to install dependency by myself but also failed.
Anyone can help?
A declarative, efficient, and flexible JavaScript library for building user interfaces.
🖖 Vue.js is a progressive, incrementally-adoptable JavaScript framework for building UI on the web.
TypeScript is a superset of JavaScript that compiles to clean JavaScript output.
An Open Source Machine Learning Framework for Everyone
The Web framework for perfectionists with deadlines.
A PHP framework for web artisans
Bring data to life with SVG, Canvas and HTML. 📊📈🎉
JavaScript (JS) is a lightweight interpreted programming language with first-class functions.
Some thing interesting about web. New door for the world.
A server is a program made to process requests and deliver data to clients.
Machine learning is a way of modeling and interpreting data that allows a piece of software to respond intelligently.
Some thing interesting about visualization, use data art
Some thing interesting about game, make everyone happy.
We are working to build community through open source technology. NB: members must have two-factor auth.
Open source projects and samples from Microsoft.
Google ❤️ Open Source for everyone.
Alibaba Open Source for everyone
Data-Driven Documents codes.
China tencent open source team.